
激素訊息傳遞與受體機制:從濃度訊號到細胞決策
受體動力學、第二訊使者級聯與訊號放大的分子與量化解析
從濃度到細胞決策:激素如何被「讀取」
一個內分泌細胞分泌的激素,血中濃度往往低到 $10^{-9}$ 至 $10^{-12}\,\text{M}$(奈莫耳至皮莫耳級)。然而它卻能在數秒內改變遠端細胞的代謝、基因表現甚至命運。這之間的關鍵,不是激素本身做了什麼,而是受體如何把一個微弱的化學濃度訊號「翻譯」成放大數百倍的細胞內反應。要理解內分泌恆定,就必須把激素—受體互動當成一個可量化的分子計算系統來看。
激素訊息傳遞的核心問題有三層:受體如何以高親和力與專一性辨識配體?訊號如何被放大與整形?以及細胞如何在持續刺激下避免飽和、維持動態範圍?本篇將從受體分類、結合動力學、第二訊使者級聯到去敏感化,逐層拆解這套機制。
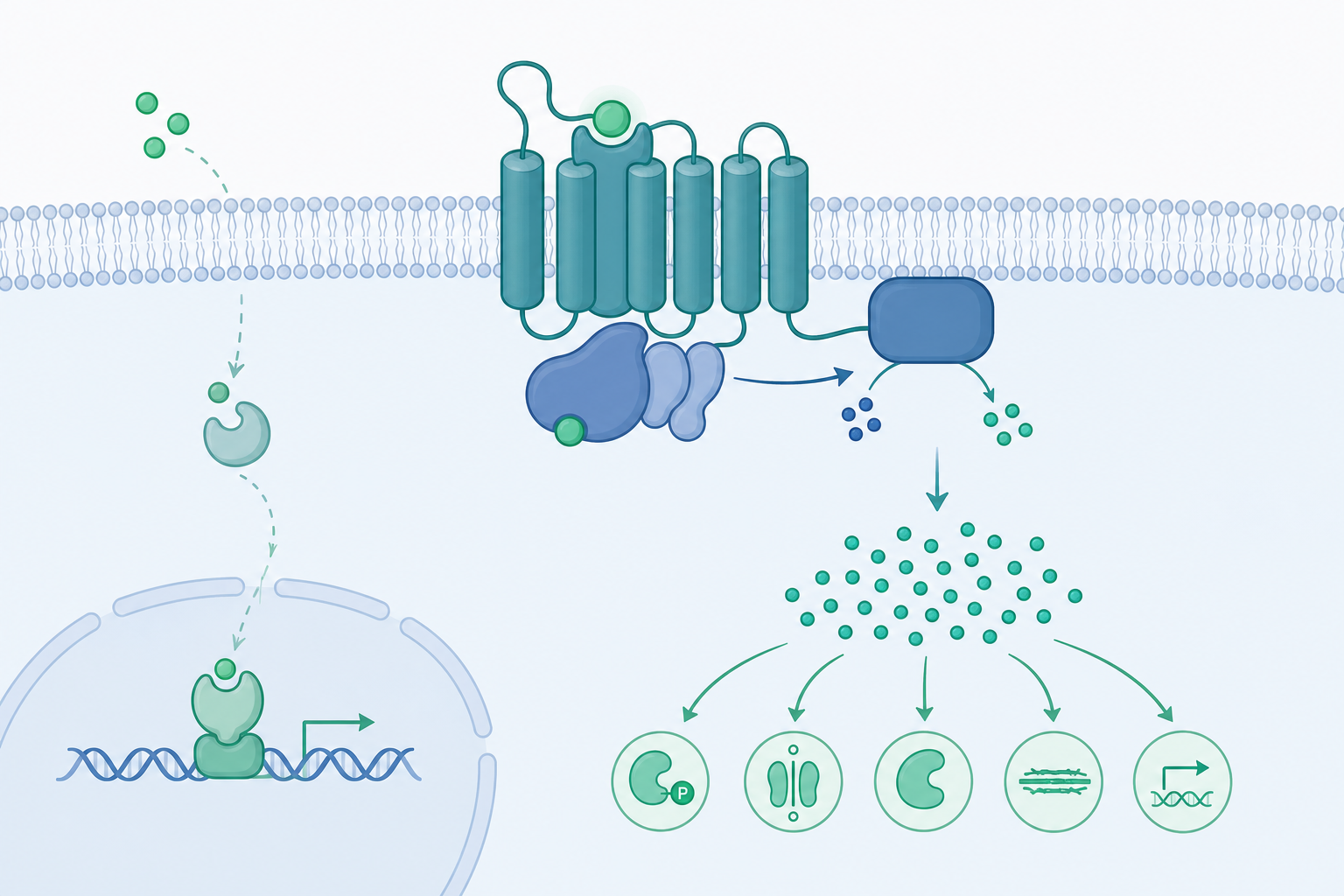
受體分類:親脂性與親水性的分流
激素的化學性質決定了受體位置。類固醇激素(如皮質醇、雌二醇)與甲狀腺素為親脂性,能穿越細胞膜,與胞內核受體結合。結合後受體—配體複合物作為轉錄因子,直接結合 DNA 上的激素反應元件(HRE),調控基因表現。這條路徑反應慢(數小時),但效果持久,本質上是改變細胞的蛋白質組成。
親水性激素(如腎上腺素、胰島素、多數胜肽激素)無法穿膜,只能作用於細胞膜受體。其中最重要的兩大類為:
- G 蛋白偶聯受體(GPCR):七次跨膜結構,配體結合後催化 G 蛋白上 GDP 換成 GTP,啟動下游酵素。人類基因組約有 800 個 GPCR,是藥物標靶最大宗。
- 受體酪胺酸激酶(RTK):如胰島素受體,配體誘導受體二聚化與自磷酸化,啟動 RAS–MAPK 與 PI3K–AKT 路徑。
結合動力學:用方程式描述親和力
受體與配體的結合可視為可逆反應:
$$R + L \rightleftharpoons RL$$
平衡時,受體占據率(被結合的比例)服從與酵素動力學同形的雙曲線關係:
$$\theta = \frac{[L]}{K_d + [L]}$$
其中 $K_d$(解離常數)等於使一半受體被占據時的配體濃度,數值越小代表親和力越高。胰島素受體的 $K_d$ 約落在奈莫耳級,恰好匹配生理激素濃度,使受體在生理範圍內具有最佳的濃度敏感度。
這個式子與 Michaelis–Menten 動力學的 $v = V_{max}[S]/(K_m + [S])$ 在數學上完全同構——這並非巧合,兩者都是「單一可逆結合位點」的必然結果。
定量小範例:若某 GPCR 對腎上腺素的 $K_d = 5\,\text{nM}$,當血中腎上腺素濃度為 $15\,\text{nM}$ 時,受體占據率為
$$\theta = \frac{15}{5 + 15} = \frac{15}{20} = 0.75$$
即 75% 的受體被占據。注意:由於下游有訊號放大,最大生理反應往往在遠低於 100% 占據率時就達成——這個現象稱為「備用受體(spare receptors)」,是內分泌系統提高靈敏度的策略之一。
第二訊使者與訊號放大
GPCR 的經典範例是腎上腺素 → β-腎上腺素受體 → $G_s$ 蛋白 → 腺苷酸環化酶(adenylyl cyclase)→ cAMP → 蛋白激酶 A(PKA)。這條級聯的威力來自催化性放大:
- 一個被占據的受體可活化多個 G 蛋白;
- 一個腺苷酸環化酶可在被活化期間產生數百個 cAMP 分子;
- 一個 PKA 可磷酸化大量下游受質。
若每一階段平均放大 100 倍,三階串聯理論上可達 $100^3 = 10^6$ 倍的放大。這解釋了為何皮莫耳級的激素能引發顯著的代謝變化(如肝醣分解)。
另一條主要路徑是 $G_q$ → 磷脂酶 C(PLC)→ 將 PIP$_2$ 切成 IP$_3$ 與 DAG。IP$_3$ 開啟內質網鈣通道,使胞質 $\text{Ca}^{2+}$ 濃度從靜息的約 $100\,\text{nM}$ 瞬間躍升十倍以上,$\text{Ca}^{2+}$ 本身即是強力的第二訊使者。
去敏感化與動態範圍
持續暴露於激素會使反應減弱,這是恆定系統的必要設計。以 GPCR 為例,去敏感化包含:
- 受體磷酸化:GPCR 激酶(GRK)磷酸化被活化的受體,招募 β-arrestin,物理上阻斷 G 蛋白偶聯;
- 受體內吞:β-arrestin 引導受體進入胞內囊泡,暫時下調表面受體數量;
- 下調:長期刺激降低受體基因表現。
去敏感化使細胞對的是「濃度變化」而非「絕對濃度」做出反應,這正是恆定回饋系統得以在寬廣背景濃度下保持靈敏的數學基礎——類似於工程上的「自適應增益控制」。
深入探討(研究所視角)
GPCR 的結構生物學革命。 過去十餘年,GPCR 從「難結晶的膜蛋白」變成結構生物學的核心戰場。Brian Kobilka 團隊於 2011 年解出 β$_2$-腎上腺素受體與其異源三聚體 $G_s$ 蛋白的活化態複合物結構(2012 年諾貝爾化學獎部分基礎),首次在原子層級展示配體結合如何透過第六跨膜螺旋(TM6)的向外擺動,打開胞內口袋容納 G 蛋白的 α5 螺旋。近年低溫電子顯微鏡(cryo-EM)的解析度革命,更使大量 GPCR–G 蛋白與 GPCR–arrestin 複合物得以在近原子解析度下被觀察,揭示了同一受體可採取多種活化構象的「構象集合(conformational ensemble)」圖像。
偏向性訊號傳遞(biased signaling)。 這項結構洞見直接催生了藥理學的範式轉移:同一受體的不同配體可以「偏向」啟動 G 蛋白路徑或 β-arrestin 路徑。傳統上 arrestin 只被視為「關閉訊號」的角色,但現已知 arrestin 本身能啟動獨立的 MAPK 級聯。偏向性配體(biased ligand)的治療意義在於:理論上可保留藥物的療效路徑、避開引發副作用的路徑。鴉片類受體的偏向性激動劑曾被寄望能止痛而不抑制呼吸,雖然臨床驗證仍具爭議,但這條設計思路已成為新藥開發主軸。
系統生物學與訊號整合。 單一路徑的線性描述無法捕捉真實細胞的行為。細胞同時接收多種激素訊號,下游路徑(如 MAPK、PI3K–AKT、cAMP)彼此交叉對話(crosstalk)並共享節點。系統生物學以常微分方程組(ODE)建模這些網路,揭示出超敏感性(ultrasensitivity)、雙穩態(bistability)與振盪等湧現性質。例如 MAPK 級聯的多階磷酸化可產生近似開關的 Hill 係數($n > 1$)反應曲線:
$$\text{response} = \frac{[S]^n}{K^n + [S]^n}$$
當 $n$ 顯著大於 1 時,系統呈現陡峭的「全有全無」切換,這對細胞命運決策(如分化與否)至關重要。
單細胞與時序動態。 傳統內分泌學測量的是細胞群體的平均反應,但單細胞活細胞影像(如 cAMP 與 $\text{Ca}^{2+}$ 的 FRET 生物感測器、ERK 核轉位報告子)揭示了被群體平均掩蓋的關鍵資訊:許多訊號是以頻率編碼(frequency encoding)而非振幅編碼傳遞的。$\text{Ca}^{2+}$ 振盪的頻率、ERK 活性的脈衝模式,都被證實攜帶不同的下游指令。這意味著激素訊息的「資訊內容」不只在於多強,更在於它的時間動態結構——這是當代內分泌訊號研究與資訊理論交會的前沿。