
心臟電生理與血壓的恆定調控:從離子梯度到壓力反射
用能斯特方程式、Frank–Starling 機制與負回饋環路,解析心臟如何把血壓鎖在恆定區間
從一個離子梯度說起:心臟為何能自己跳
把一顆離體的心臟泡在含氧的生理鹽水裡,它仍會規律地搏動——這個現象在十九世紀就讓生理學家著迷。心臟的自律性(automaticity)不需要大腦下令,而是源自竇房結(sinoatrial node, SA node)細胞膜上一組離子通道的時序協奏。要理解血壓如何被維持在一個狹窄的恆定區間,就必須先把鏡頭拉到單一細胞的膜電位變化,再逐層放大到整體循環的回饋網路。本文聚焦於三個彼此咬合的層次:心肌細胞的電生理、心輸出量與外周阻力的力學決定因子,以及神經–體液系統如何把平均動脈壓鎖定在約 90–100 mmHg。
膜電位的根本來自跨膜離子的電化學梯度。對單一離子種類 $X$,其平衡電位由能斯特方程式(Nernst equation)給出:
$$E_X = \frac{RT}{zF}\ln\frac{[X]_o}{[X]_i}$$
其中 $R$ 為氣體常數、$T$ 為絕對溫度、$z$ 為離子價數、$F$ 為法拉第常數。在 37 ℃ 下,$\frac{RT}{F}\approx 26.7\ \text{mV}$,換成以 10 為底的對數約為 $61.5\ \text{mV}$。以心肌細胞 $[K^+]_i\approx 140\ \text{mM}$、$[K^+]_o\approx 4\ \text{mM}$ 代入:
$$E_K = 61.5\times\log_{10}\frac{4}{140}\approx -95\ \text{mV}$$
這個 $-95\ \text{mV}$ 正是工作型心肌靜息電位接近的數值,也說明了為何高血鉀($[K^+]_o$ 上升)會使 $E_K$ 去極化、進而引發致命性心律不整——它直接動搖了膜電位的化學根基。
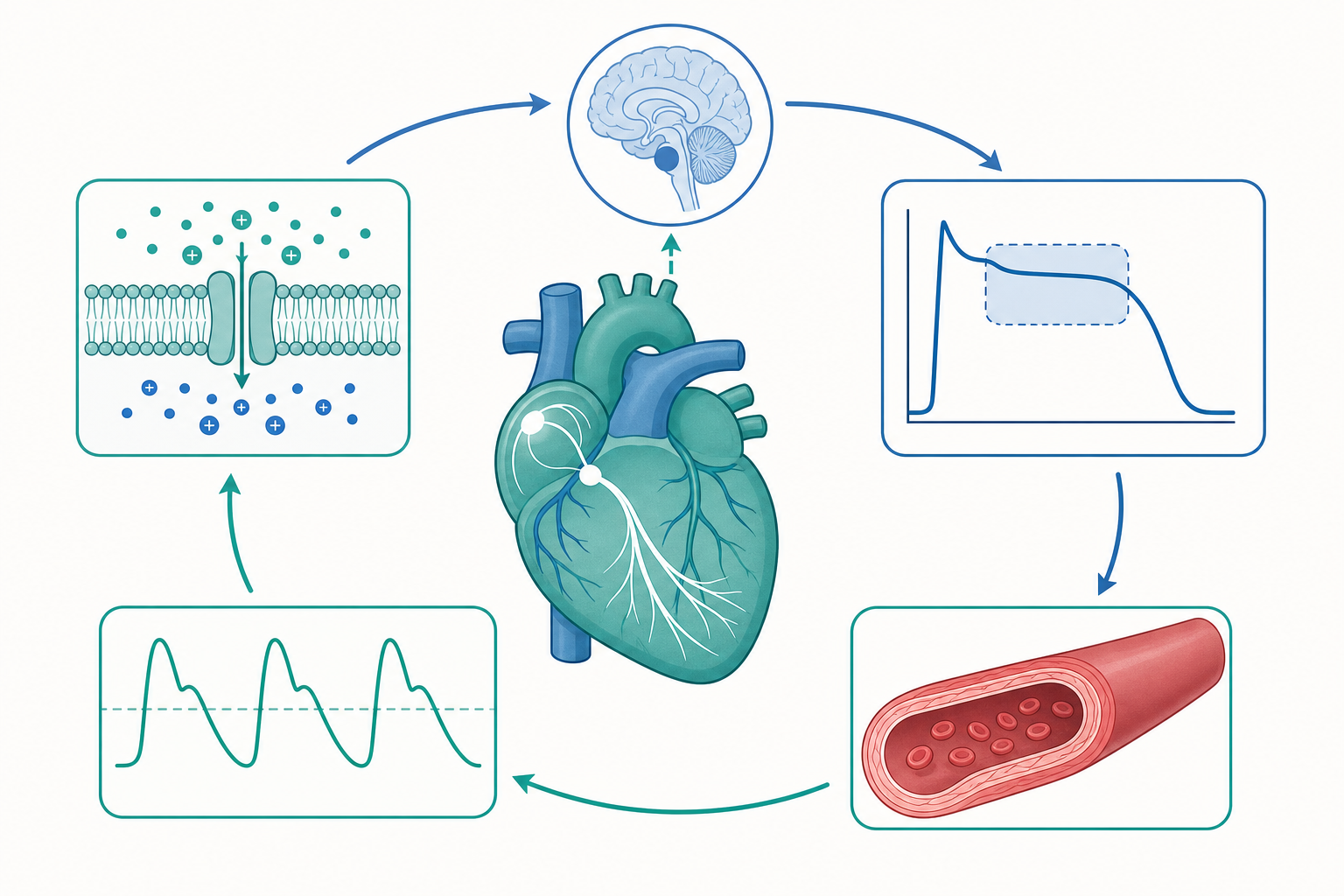
動作電位的離子分工:起搏細胞與工作心肌的差異
竇房結細胞沒有穩定的靜息電位,而是有一段緩慢的舒張期去極化(phase 4),這正是心律的「節拍器」。其關鍵推手是超極化活化的 $I_f$「funny current」(由 HCN 通道介導,對 $Na^+$ 與 $K^+$ 皆通透),加上 T 型與 L 型鈣電流的接力。當膜電位漂移到約 $-40\ \text{mV}$ 的閾值,L 型鈣通道(Ca$_v$1.2)大量開啟,產生以鈣為主的緩慢上升相——這與工作心肌靠快速 $Na^+$ 內流(Na$_v$1.5)的尖銳 phase 0 截然不同。
工作型心室肌的動作電位有著教科書式的五個時相:phase 0 快速去極化($I_{Na}$)、phase 1 短暫複極($I_{to}$)、phase 2 平台期($I_{Ca,L}$ 內流與 $I_{K}$ 外流達成微妙平衡)、phase 3 複極(延遲整流鉀電流 $I_{Kr}$、$I_{Ks}$)、phase 4 靜息(內向整流 $I_{K1}$ 維持)。長達約 200–300 ms 的平台期造就了心肌的長不應期,使其無法像骨骼肌那樣產生強直收縮——這是心臟作為節律性幫浦的安全機制。
平台期的鈣內流還觸發了「鈣誘發鈣釋放」(calcium-induced calcium release, CICR):少量經 L 型通道進入的 $Ca^{2+}$ 打開肌漿網上的 ryanodine 受體(RyR2),釋放大量儲存鈣,啟動肌絲滑動。收縮後,鈣須被 SERCA2a 泵回肌漿網、並由 Na$^+$/Ca$^{2+}$ 交換子(NCX)排出細胞,舒張才能完成。心衰竭中常見的 SERCA2a 表現下降,正是舒張功能障礙的分子註腳。
從單細胞到整體:心輸出量與血壓的力學
整體血壓並非由電生理單獨決定,而是循環力學的乘積。平均動脈壓(mean arterial pressure, MAP)可近似為:
$$\text{MAP} \approx \text{CO} \times \text{TPR}$$
其中心輸出量 $\text{CO} = \text{HR} \times \text{SV}$(心率乘以每搏輸出量),TPR 為總外周阻力。每搏輸出量由前負荷、後負荷與收縮力三者決定,而前負荷與心室充盈體積透過 Frank–Starling 機制連結:心肌纖維在生理範圍內被拉得越長,肌節中肌動–肌凝蛋白的有效重疊與肌鈣蛋白對鈣的敏感度越高,收縮力越強。這是心臟的「自我調節」——靜脈回流增加時,心臟自動以更強的收縮把多餘血量打出去。
外周阻力則由小動脈半徑主宰,這一點 Poiseuille 關係交代得很清楚:層流下阻力與半徑四次方成反比,
$$R = \frac{8\eta L}{\pi r^4}$$
半徑只要縮小 16%,阻力就翻倍。這解釋了為何血管平滑肌張力的微調(受交感神經、血管收縮素 II、一氧化氮等共同影響)能對血壓產生槓桿般的放大效果。
恆定的守門人:壓力反射與神經–體液回饋
血壓的瞬時穩定靠的是壓力反射(baroreflex)。頸動脈竇與主動脈弓的機械敏感壓力受器偵測管壁張力,經舌咽神經與迷走神經把訊號送到延腦孤束核。當血壓上升,傳入放電增加,中樞反射性增強迷走(副交感)輸出、抑制交感輸出,使心率下降、血管舒張、血壓回落;反之亦然。這是一個負回饋環路,時間常數在秒級。
長時程的血壓校準則交給腎臟主導的腎素–血管收縮素–醛固酮系統(RAAS)。腎血流灌注下降時,腎絲球旁器釋出腎素,啟動血管收縮素原 → 血管收縮素 I →(經 ACE)→ 血管收縮素 II 的級聯,後者既直接收縮血管,又刺激醛固酮促進鈉水滯留、擴增血容量。腎臟的「壓力–利尿」(pressure-natriuresis)特性——血壓越高、排鈉越多——構成血壓的長期設定點,也是多數降壓藥(ACE 抑制劑、ARB、利尿劑)的作用靶點。
定量小範例:壓力反射的增益
假設某人基礎 MAP 為 100 mmHg,因急性失血使 MAP 瞬間掉到 70 mmHg。若壓力反射的開環增益(open-loop gain)$G = 5$,定義為「校正量 / 殘餘誤差」,則閉環後殘餘的壓力偏差為:
$$\Delta P_{\text{closed}} = \frac{\Delta P_{\text{open}}}{1+G} = \frac{-30}{1+5} = -5\ \text{mmHg}$$
也就是說,反射把原本 $-30\ \text{mmHg}$ 的擾動壓縮到只剩約 $-5\ \text{mmHg}$,校正了約 83%。增益越高,系統把血壓「拉回設定點」的能力越強——這也是為何壓力反射敏感度(baroreflex sensitivity, BRS)下降被視為心血管事件的獨立危險因子。
深入探討(研究所視角)
把上述機制推到最前沿,三條主線值得關注。
其一,離子通道的結構生物學與致心律不整的精準對應。 近十餘年低溫電子顯微術(cryo-EM)讓多個心臟通道的原子結構得以解析,從 Na$_v$1.5、Ca$_v$1.2 到 hERG($I_{Kr}$ 的分子基礎)。hERG 通道有一個異常寬大、富含芳香殘基的中央腔,使大量結構不相關的藥物得以結合並阻斷它,這正是「藥物誘發長 QT 症候群」的分子根源;如今藥廠在臨床前須例行做 hERG 篩選。能把點突變(如 LQT3 的 Na$_v$1.5 ΔKPQ 缺失導致 late $I_{Na}$ 增大)對應到具體的閘控缺陷與心電圖延長,是離子通道病(channelopathy)研究的典範。
其二,鈣處理的奈米域(nanodomain)與興奮–收縮耦聯的時空精度。 RyR2 與 L 型鈣通道在 T 小管–肌漿網接面組成「couplon」,兩者間僅約 12–15 nm 的裂隙構成功能性奈米域,使 CICR 具備局部、全有全無的「鈣火花」(calcium spark)特性。超解析顯微術(STORM/DNA-PAINT)已能解析單一 couplon 內 RyR2 的數目與排列;在心衰與心房顫動模型中,CaMKII 過度磷酸化 RyR2 造成舒張期鈣漏(diastolic Ca leak),既損害收縮也誘發延遲後去極化(DAD)這類觸發活動。把分子尺度的鈣漏連到組織尺度的折返性心律不整,是目前計算–實驗整合最活躍的戰場之一。
其三,多尺度系統生物學模型與單細胞體學。 自 Noble 於 1960 年代建立首個心肌動作電位數學模型以來,O'Hara–Rudy 等當代人類心室肌模型已能把數十條離子電流耦合進可模擬藥物效應的虛擬心臟,FDA 的 CiPA 倡議甚至把這類 in silico 模型納入藥物致心律不整風險評估的監管框架。與此同時,單細胞 RNA 定序揭示了竇房結與傳導系統細胞的異質性,重新定義了「起搏細胞」並非單一群體;空間轉錄體學則開始描繪傳導系統在三維心臟中的分子地形。把能斯特方程式這層百年老物理,一路接到 cryo-EM 結構、奈米域動力學與多體學資料,正是當代心臟生理「從分子到器官」整合研究的縮影——而血壓恆定,不過是這套精密系統最終呈現於床邊血壓計上的一個讀數。