
分子親緣分析與系統發生樹:從序列差異到演化時間軸
替換模型、最大概似、分子鐘與物種樹重建的量化內核
從序列差異讀出演化的時間軸
當我們把人類、黑猩猩與小鼠的某段基因排在一起,會看到一連串「相同」與「相異」的位點。直覺上,相異越多代表親緣越遠——但這只是起點。分子親緣分析(molecular phylogenetics)真正要做的,是把這些離散的字元差異,轉化為一棵帶有分支拓樸、分支長度,甚至絕對年代的系統發生樹(phylogenetic tree)。要做到這件事,必須同時面對三個層次的問題:突變如何在分子層次累積、如何用機率模型描述這個累積過程,以及如何從龐大的樹空間中選出最可信的那一棵。
本篇假設你已熟悉中心法則、點突變類型與基本族群遺傳概念,接下來直接進入機制與量化。
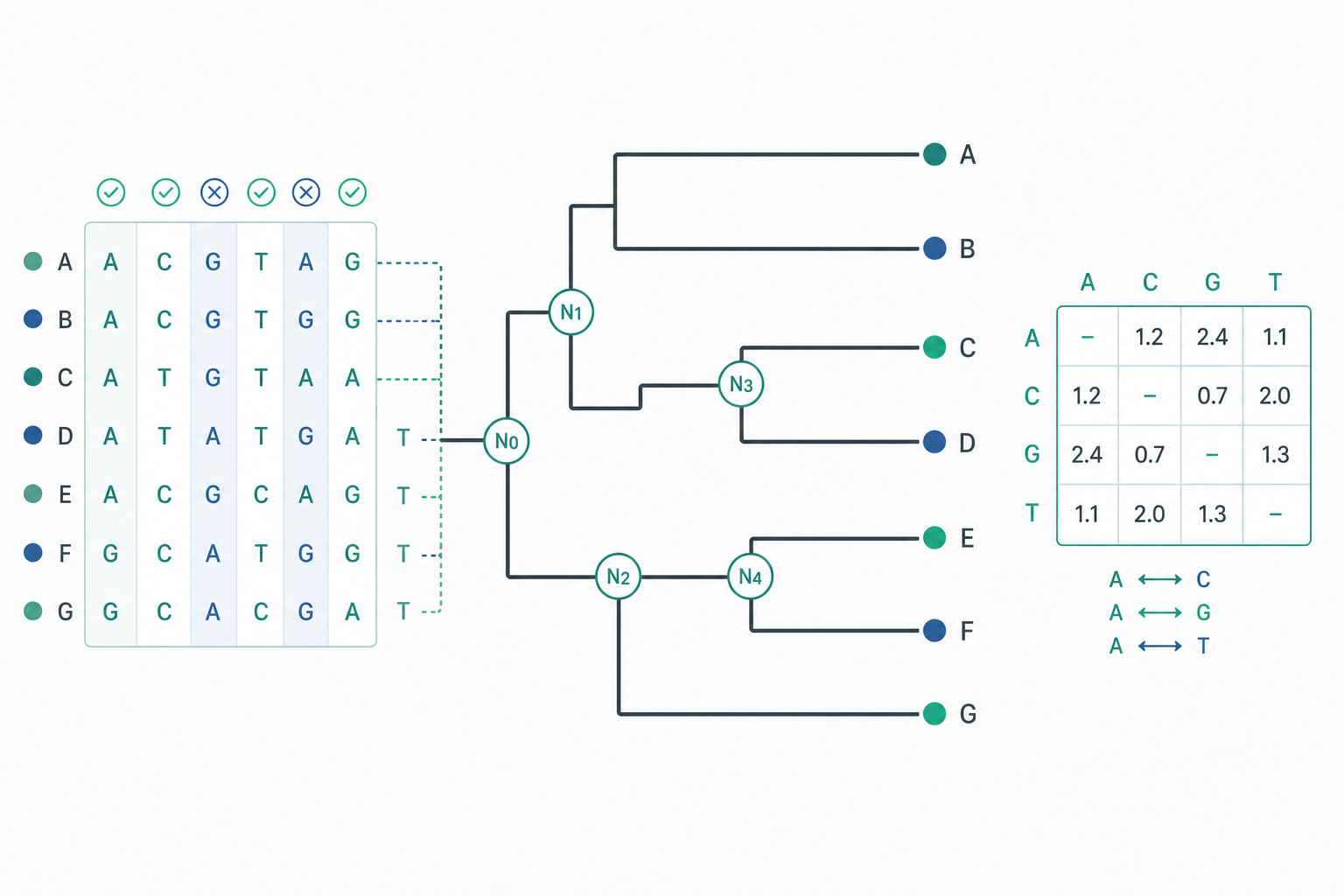
為什麼觀察到的差異會「飽和」
最樸素的距離度量是 $p$-distance,也就是兩條序列中相異位點的比例。問題在於:同一個位點可能發生多次替換(multiple hits),例如 A→G→A 會讓我們「看不到」實際發生過兩次事件。因此觀察距離 $p$ 會系統性地低估真實的替換數,且在演化時間拉長時逐漸飽和(saturation)。
最簡單的校正是 Jukes–Cantor(JC69)模型,它假設四種核苷酸彼此互換的速率都相同。在此假設下,校正後的每位點替換數為:
$$d = -\frac{3}{4}\ln\!\left(1 - \frac{4}{3}p\right)$$
當 $p$ 很小時 $d \approx p$;但當 $p$ 接近 $0.75$(四種鹼基隨機分布的期望差異)時,$d$ 趨於無窮大——這正是「飽和」的數學表現。更精緻的模型(如 Kimura 2-parameter 區分轉換 transition 與顛換 transversion,HKY85、GTR 進一步放寬鹼基頻率與各類替換速率)都建立在同一個核心觀念上:用一個連續時間馬可夫過程描述替換,並從速率矩陣 $Q$ 推得任意時間 $t$ 後的替換機率矩陣 $P(t)=e^{Qt}$。
從距離到樹:四種建樹典範
距離法(如 Neighbor-Joining)先把所有兩兩校正距離填進矩陣,再以最小化整體分支長度的準則逐步聚合。它運算極快、適合上萬序列的初篩,但把序列壓縮成單一數字會丟失位點層次的資訊。
最大簡約法(Maximum Parsimony)尋找「所需替換事件總數最少」的樹。它直覺、無需模型,但在分支長度差異懸殊時會落入長枝吸引(long-branch attraction)——兩條快速演化的長枝因為各自累積大量隨機相似,被錯誤地聚在一起。
最大概似法(Maximum Likelihood, ML)與貝氏推論(Bayesian inference)是當代主流。ML 在給定替換模型下,尋找使資料概似 $L=P(\text{data}\mid \text{tree},\,\theta)$ 最大的樹與參數;貝氏法則透過 MCMC(如 MrBayes、BEAST 採用的 Metropolis-coupled 演算法)對樹空間採樣,輸出每個分支的後驗機率。兩者的計算核心都是 Felsenstein 的剪枝演算法(pruning algorithm),它以動態規劃在樹上由葉向根遞迴計算條件概似,把原本指數級的祖先狀態加總壓到線性時間。
速率異質性與分子鐘
真實序列中,不同位點的演化速率天差地別:密碼子第三位、非編碼區變化快,功能關鍵殘基幾乎不動。忽略這點會嚴重低估深層分歧。標準做法是引入跨位點的 Γ 分布速率異質性(常記為 +Γ 或 +G),把速率切成數個離散類別積分。
要把分支長度(以替換數計)換算成絕對年代,需要分子鐘假設。嚴格分子鐘假設所有譜系速率恆定,但這在跨度大的資料中常被推翻;現代多採鬆弛分子鐘(relaxed clock),讓速率沿樹獨立或自相關地變動,再用化石或地質事件作為校準點(calibration)轉換為時間。
定量小範例:JC69 距離校正
假設兩條同源 DNA 序列長 $300$ 個位點,比對後發現 $45$ 個位點相異。
第一步,計算觀察距離:
$$p = \frac{45}{300} = 0.15$$
第二步,代入 JC69 公式校正多重替換:
$$d = -\frac{3}{4}\ln\!\left(1 - \frac{4}{3}\times 0.15\right) = -0.75\,\ln(0.80)$$
由於 $\ln(0.80)\approx -0.2231$:
$$d \approx -0.75 \times (-0.2231) \approx 0.167\ \text{替換/位點}$$
可見校正後的 $0.167$ 高於未校正的 $0.15$,差距約 $11\%$,正反映被「看不見的」多重替換所掩蓋的事件。若再假設分子鐘速率為每位點每年 $r = 1\times 10^{-9}$、且 $d$ 是兩譜系自共同祖先以來各自累積的總和,則分歧時間約為:
$$T = \frac{d}{2r} = \frac{0.167}{2\times 10^{-9}} \approx 8.3\times 10^{7}\ \text{年}$$
也就是約 $8{,}300$ 萬年前分歧——這正是分子鐘把序列差異轉成地質時間軸的具體運算。
不只一棵基因樹:物種樹的挑戰
一個常見誤解是「基因樹等於物種樹」。事實上,受不完全譜系篩選(incomplete lineage sorting, ILS)、基因水平轉移與基因重複影響,不同基因常給出彼此衝突的拓樸。當分歧事件密集發生(短內部分支),祖先族群中的多型性可能跨越物種形成事件而隨機固定,導致個別基因樹與真實物種樹不一致。多基因座(multilocus)資料與合併模型(如 multispecies coalescent,ASTRAL、*BEAST 所實作)正是為了在這種雜訊下重建可信的物種樹。自助法(bootstrap)重抽位點、以及貝氏後驗機率,則用來量化每個分支的支持度——一個拓樸正確但支持度低的分支,在生物學詮釋上仍須保留。
深入探討(研究所視角)
在前沿研究中,分子親緣分析正與結構生物學、體學與系統生物學深度交織,呈現主體未觸及的幾個張力與機會。
結構約束下的替換模型。 標準 GTR+Γ 假設所有位點獨立演化,但蛋白質殘基間存在強烈的共演化(coevolution):維持折疊與結合界面的成對殘基必須協同變動。直接耦合分析(Direct Coupling Analysis, DCA)以最大熵模型從多序列比對推斷殘基對之間的統計耦合,這些訊號不僅用於預測接觸圖(contact map,AlphaFold 之前的結構預測主力之一),也揭示位點獨立假設的系統性偏誤。當我們把結構穩定性(如 $\Delta\Delta G$ 折疊自由能變化)納入適應度地形,替換速率便不再是常數,而是位點所處結構與功能脈絡的函數——這催生了結構感知(structure-aware)系統發生模型與突變-選擇(mutation–selection)框架,後者明確分離突變偏好與選擇係數。
體學尺度的譜系重建。 系統發生學的對象已從單基因擴展到全基因組。系統發生基因體學(phylogenomics)動輒處理上千個基因座的串接或合併分析,此時模型誤設(model misspecification)與不同基因的歷史衝突成為核心議題;組成異質性(compositional heterogeneity)需要 CAT 等混合模型來吸收跨譜系的鹼基/胺基酸頻率變化,否則易產生長枝吸引假象。在更細的尺度上,單細胞譜系追蹤(single-cell lineage tracing)把同樣的樹思維帶進個體發育與腫瘤演化:CRISPR 誘導的可遺傳條碼或自然體細胞突變,讓研究者得以重建細胞的分裂歷史,繪製腫瘤內亞克隆(subclone)的演化樹,並推斷轉移事件的方向——這在概念上與物種樹重建同構,卻面臨突變率漂移與資料缺失的全新挑戰。
系統生物學與選擇訊號的量化。 把演化動力學寫成可檢驗的統計模型,是當代的核心工作。$dN/dS$(非同義對同義替換比,常記為 $\omega$)是偵測正選擇的經典量:$\omega>1$ 暗示適應性演化,$\omega<1$ 暗示純化選擇。分支-位點模型(branch-site model)進一步允許特定譜系的特定位點短暫進入 $\omega>1$ 狀態,用來定位免疫逃逸或宿主適應等局部選擇事件——這在病毒(如流感 HA 抗原位點、SARS-CoV-2 棘蛋白)的即時演化監測中已成標準工具。值得強調的是,這類推論本質上是統計假說檢定,需以概似比檢定(LRT)對抗虛無模型,並警覺於重組與比對誤差造成的偽陽性。
時間維度與生死過程模型。 把譜系本身視為一個隨機過程,是 macroevolution 的重要進展。出生-死亡模型(birth–death process)與譜系合併理論(coalescent)為樹的形狀提供生成式先驗,使我們能從現存物種的樹推斷過去的多樣化速率變化,甚至偵測大滅絕的速率轉折(如 BAMM、出生-死亡-取樣模型)。而化石化生死過程(fossilized birth–death)更把化石直接當作樹上的取樣節點,統一了形態與分子、滅絕與現存的證據,讓校準不再依賴少數武斷的化石點。這些方法的共同精神,是把「樹」從一個被動的重建結果,提升為一個承載演化過程參數、可被資料反推與檢定的動態模型。