
連鎖、互換與染色體作圖:從重組頻率到分子重組景觀
用重組頻率、作圖函數與 LOD score 重建染色體上的基因地圖
從「不獨立分配」說起:當基因住在同一條染色體上
孟德爾的自由分配律告訴我們,不同性狀的等位基因會獨立分配。但這條定律有個前提:被觀察的基因要位在不同對的染色體上。當兩個基因座緊鄰在同一條染色體上時,它們傾向「綁在一起」遺傳——這就是連鎖(linkage)。連鎖打破了 9:3:3:1 的期望比,卻也意外地給了我們一把尺:透過量化兩基因「被拆開」的頻率,我們可以重建基因在染色體上的線性排列順序與相對距離。這一節之後,我們會看到連鎖、互換與作圖如何構成一個自洽的量化體系。
互換的分子機制:減數分裂中的程序性 DNA 斷裂
連鎖之所以「不完美」,是因為減數分裂前期 I 會發生互換(crossing over)。在偶線期到粗線期,同源染色體配對形成聯會複合體(synaptonemal complex),而互換的起點是一個受嚴格調控的雙股斷裂(DSB)。在多數真核生物中,這個斷裂由 Spo11 拓撲異構酶樣蛋白以共價方式切開 DNA,隨後 MRN 複合體切除 Spo11 留下短的 oligonucleotide,露出 3′ 單股尾端。
接著 Dmc1/Rad51 重組酶在單股 DNA 上組裝成核蛋白絲,進行同源序列搜尋與股入侵,形成 D-loop。若依循 DSBR 路徑,會生成雙 Holliday junction,其解離方式(resolution)決定了結果是交叉型(crossover)還是非交叉型(non-crossover)。只有交叉型互換會造成側翼遺傳標記的重組,也就是我們在作圖時量測的事件。
值得強調的是互換干涉(interference):一個互換的發生會抑制鄰近區域再發生互換,使得交叉點沿染色體的分布比隨機更均勻。我們用并發係數(coefficient of coincidence, $c$)量化它:
$$c = \frac{\text{觀測雙互換頻率}}{\text{期望雙互換頻率}}, \qquad I = 1 - c$$
其中 $I$ 為干涉值。$I=1$ 表示完全干涉(觀測不到雙互換),$I=0$ 表示互換彼此獨立。
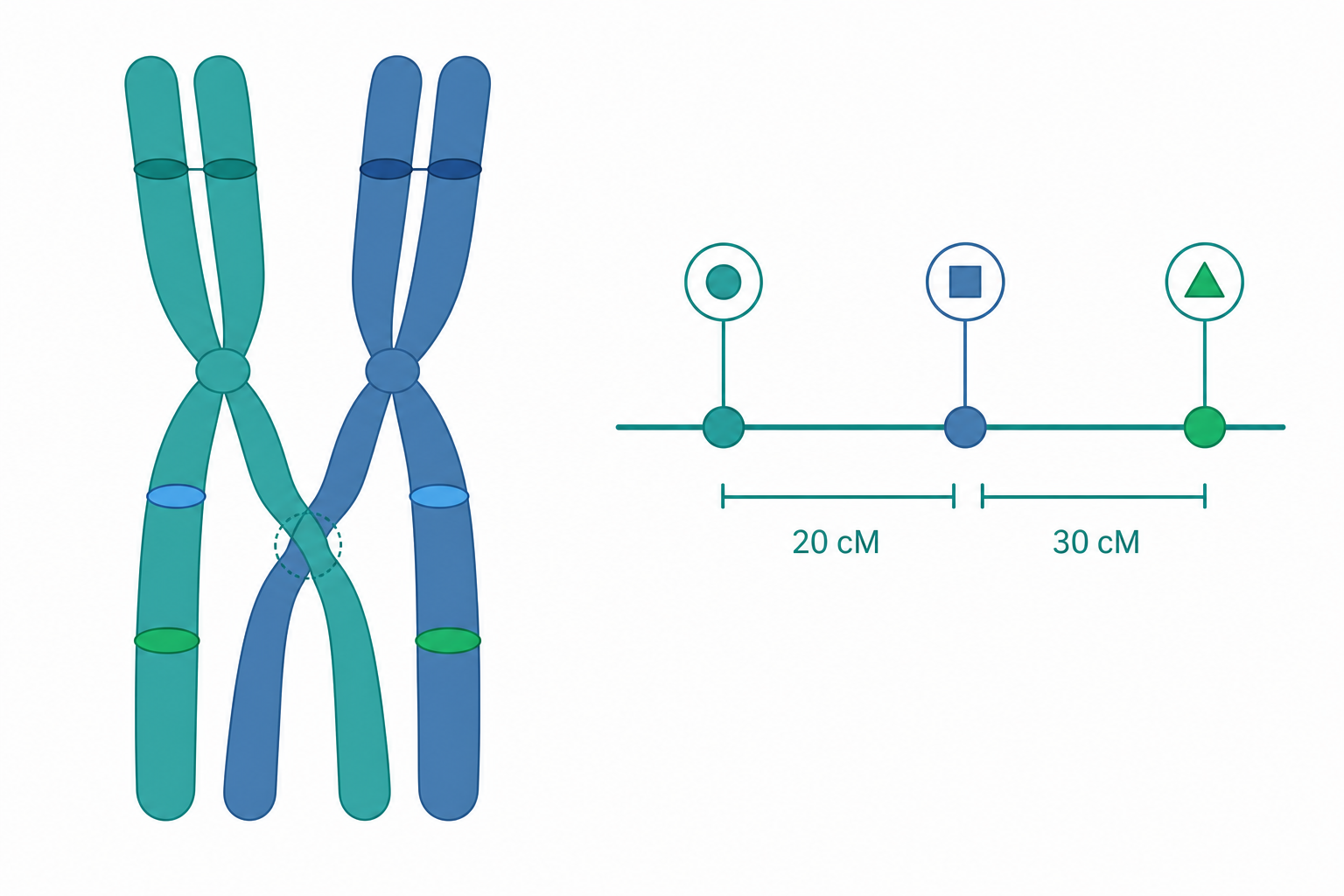
重組頻率作為遺傳距離:圖距單位與三點測交
連鎖分析的核心量是重組頻率(recombination frequency, RF):
$$RF = \frac{\text{重組型後代數}}{\text{總後代數}}$$
Sturtevant 在 1913 年的洞見是:兩基因在染色體上相距越遠,中間發生互換的機會越大,$RF$ 越高。於是定義 1 圖距單位(centimorgan, cM)對應 1% 的重組頻率。然而 $RF$ 並非無上限——它最高趨近 0.5(等同於獨立分配),因為距離夠遠時幾乎每個減數分裂都會發生奇數次互換。這意味著對遠距離基因,$RF$ 會飽和並低估真實的物理/圖距離。
要在多基因情境下確立順序與校正距離,三點測交(three-point testcross)是經典工具。以三個連鎖基因 $A$、$B$、$C$ 為例,雜合三重雜合子 $\times$ 三隱性個體的後代會出現 8 種表現型,可分為親本型(最多)、單互換型兩類、與雙互換型(最少)。雙互換型相對於親本型,只有「中間那個基因」的等位被交換,據此即可判定基因順序。
定量小範例:設三點測交 1000 個後代,區間 $A$–$B$ 的單互換 80 個、$B$–$C$ 的單互換 120 個、$A$–$C$ 的雙互換 10 個。
各區間距離(雙互換同時影響兩側,須計入兩次):
$$d_{AB} = \frac{80 + 10}{1000} \times 100 = 9.0\ \text{cM}$$
$$d_{BC} = \frac{120 + 10}{1000} \times 100 = 13.0\ \text{cM}$$
期望雙互換頻率 $= 0.09 \times 0.13 = 0.0117$,觀測 $= 10/1000 = 0.010$,故
$$c = \frac{0.010}{0.0117} \approx 0.855, \qquad I = 1 - 0.855 \approx 0.145$$
即此區間存在約 15% 的干涉,雙互換略少於隨機期望。
把 RF 換算成真正的圖距:作圖函數
因為遠距離下會發生多重互換(其中偶數次互換不產生可觀測的重組),$RF$ 與真實圖距 $m$(以 Morgan 計,期望互換次數)之間是非線性關係。若假設互換沿染色體服從 Poisson 分布且無干涉,Haldane 作圖函數給出:
$$RF = \frac{1}{2}\left(1 - e^{-2m}\right)$$
反解可由觀測 $RF$ 估計真實圖距 $m = -\tfrac{1}{2}\ln(1 - 2\,RF)$。當 $RF$ 很小時,泰勒展開 $RF \approx m$,回到「1 cM ≈ 1%」的近似;但 $RF$ 接近 0.5 時,$m$ 會發散,正反映距離資訊的飽和。
若要納入干涉,Kosambi 作圖函數更貼近真實生物資料:
$$RF = \frac{1}{2}\tanh(2m)$$
它在短距離與 Haldane 一致,但在中長距離因隱含干涉而給出較保守的距離估計。這也是為何當代連鎖圖譜多採 Kosambi 距離。
從遺傳圖到物理圖:分子標記與 LOD score
古典作圖依賴可見表現型,現代連鎖分析則改用 SNP、微衛星等分子標記,可在全基因組密集布點。對於人類等無法做測交的物種,我們用對數似然比(LOD score)評估連鎖:
$$\text{LOD} = \log_{10}\frac{L(\theta)}{L(\theta = 0.5)}$$
其中 $\theta$ 為重組分數,$L$ 為對應的似然函數。慣例上 $\text{LOD} \geq 3$(即連鎖假說的可能性是無連鎖的 1000 倍以上)視為顯著連鎖,$\leq -2$ 則排除連鎖。這套方法曾在定位囊腫性纖維化、亨丁頓舞蹈症等致病基因上扮演關鍵角色。
深入探討(研究所視角)
連鎖與互換的當代研究已遠超「數重組型後代」的層次,進入互換熱點的表觀基因體決定論與單細胞減數分裂體學。在哺乳類中,互換並非沿染色體均勻分布,而是高度集中在數萬個「重組熱點」上。關鍵發現是鋅指蛋白 PRDM9:它以快速演化的 C2H2 鋅指陣列辨識特定 DNA 模體,並透過其 PR/SET 結構域催化 H3K4me3 與 H3K36me3 雙重組蛋白甲基化標記,將 Spo11 引導至這些位點。PRDM9 等位基因的差異會重新定義整個基因體的熱點地圖,而「熱點悖論」——被偏好的識別模體因偏向基因轉換(biased gene conversion)而自我侵蝕——正是 PRDM9 在脊椎動物中受正向選擇、快速演化的演化動力學解釋。PRDM9 還與物種形成相關:小鼠雜交不孕的部分遺傳基礎可追溯到 PRDM9 結合位點在親本間的不對稱侵蝕,導致非同源序列無法正確完成重組。
結構生物學層面,聯會複合體的橫向絲(SYCP1)與中央元件、以及交叉點上 MutLγ(MLH1–MLH3)核酸內切酶如何將 Holliday junction 偏向交叉型解離,是近年冷凍電鏡與生化重構的焦點。MLH1 灶點數目可作為交叉點的細胞學標記,使我們能在單一精母細胞層級計數互換並驗證干涉。
在量化模型上,互換干涉的機制已從唯象描述推進到物理模型。beam-film 模型將染色體軸視為承受機械應力的彈性桿,交叉點的形成釋放局部應力並沿軸傳播,抑制鄰近交叉,從而以應力鬆弛的特徵長度自然解釋干涉,並能定量預測交叉點數目與間距分布——這比 Kosambi 函數的經驗 $\tanh$ 形式提供了更深的物理基礎。此外,「交叉保證(crossover assurance)」——每對同源染色體至少需一個義務性交叉(obligate crossover)以確保正確分離,否則導致非整倍體——也被整合進這類模型,連結了減數分裂錯誤與唐氏症等臨床表型。
單細胞與單分子技術正在改寫資料尺度。sperm-typing 與單精子全基因組定序可直接觀察單一減數分裂產物的重組斷點,無需世代雜交;Hi-C 與 Strand-seq 則把減數分裂的三維染色體構象與股導向資訊納入,使「遺傳圖—物理圖—三維構象圖」得以整合。在群體尺度,連鎖不平衡(LD)衰減曲線承載了歷史重組與有效族群大小的訊息,GWAS 中的精細定位(fine-mapping)即奠基於 LD 結構。值得注意的是,性別二態性(heterochiasmy)——人類女性的全基因組圖距約為男性的 1.6 倍——以及年齡相關的重組率漂移,都顯示互換景觀是受發育與環境共同塑形的動態系統,而非固定不變的物理常數。這些都讓「染色體作圖」從一張靜態地圖,演化為一門研究基因體穩定性與演化的活躍科學。