
蛋白質的四級結構與摺疊:從能量漏斗到別構協同
以自由能、Hill 方程與摺疊漏斗,剖析多肽鏈如何組裝成具功能的分子機器
從一維序列到三維機器:摺疊問題的核心
一條由胺基酸串成的多肽鏈,本質上只是一維資訊;但它在水中往往能在毫秒到秒的尺度內,自發摺疊成唯一且具功能的三維構象。Anfinsen 在 1960 年代以核糖核酸酶的變性/復性實驗證明:天然構象的所有資訊原則上已編碼於一級序列之中——這就是「熱力學假說」。換句話說,天然態通常對應於可及構象空間中的自由能最小值。
我們可以把摺疊的驅動力寫成一個吉布斯自由能的平衡:
$$\Delta G_{\text{fold}} = \Delta H - T\Delta S$$
這裡有兩個彼此競爭的熵項。鏈本身摺疊會損失構象熵($\Delta S_{\text{chain}} < 0$,不利),但疏水側鏈的埋藏會釋放原本被「水籠」(clathrate-like cage)有序排列的水分子,貢獻一個大的正值溶劑熵——這正是疏水效應作為摺疊主要驅動力的本質。再加上氫鍵、凡得瓦力與鹽橋的焓貢獻,淨效應使天然態僅比變性態穩定約 $5\text{–}15\ \text{kcal/mol}$。這個數字驚人地小:意味著一個摺疊蛋白只比解離態「穩定幾個氫鍵」而已,邊際穩定性正是生命可調控與可降解的代價。
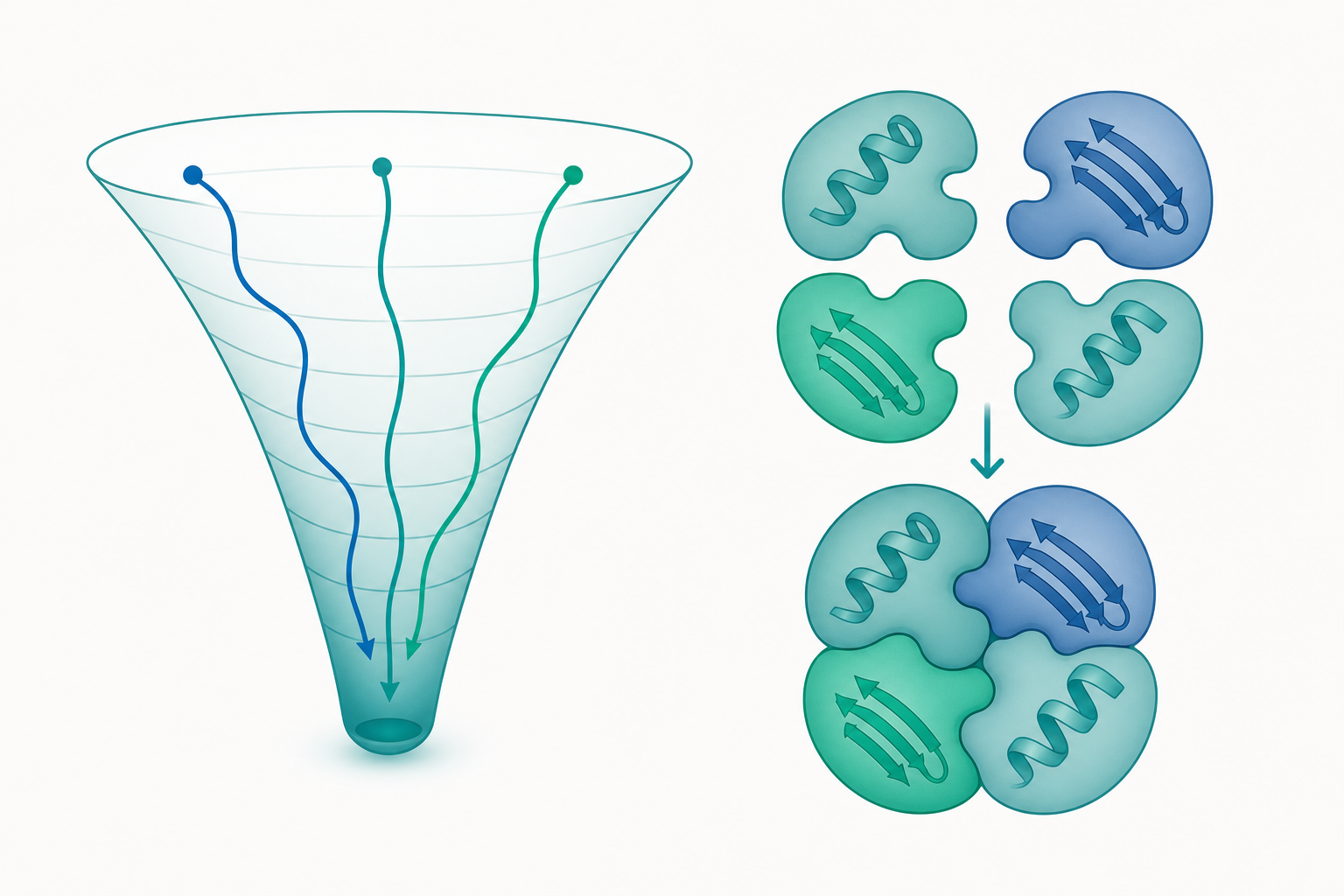
Levinthal 弔詭與摺疊漏斗
若一條 100 殘基的鏈,每個肽鍵假設只有 3 種構象,則總構象數約 $3^{100}\approx 5\times10^{47}$。即使每種構象切換僅需 $10^{-13}$ 秒,窮舉搜尋也要遠超宇宙年齡——這就是 Levinthal 弔詭。蛋白顯然不是隨機搜尋,而是沿著有偏好的路徑下降。
現代觀點以「摺疊能量漏斗」(folding funnel)取代單一路徑:構象空間像一個粗糙的漏斗,縱軸是自由能、橫向廣度代表構象熵。鏈從漏斗口(高熵、未摺疊)一路向下,多條微觀路徑殊途同歸,中途可能被局部極小(kinetic traps)暫時困住,形成摺疊中間態。漏斗的「粗糙度」決定了是否出現可偵測的中間態,而漏斗的整體傾斜度決定摺疊速率。
四級結構:當次單元組裝成功能整體
許多蛋白並非以單體行使功能,而需數條多肽鏈(次單元)以非共價作用力(與少量雙硫鍵)組裝成寡聚體,此即四級結構。四級結構帶來單體無法達成的性質:協同性(cooperativity)、別構調控(allostery)、活性位點的跨次單元構築,以及更高的結構穩定性。
血紅素是經典範例。它由 $\alpha_2\beta_2$ 四個次單元組成,氧結合呈現 S 型的協同曲線,可用 Hill 方程描述飽和分數 $Y$:
$$Y = \frac{[\text{O}_2]^{n}}{K_d + [\text{O}_2]^{n}} = \frac{(pO_2)^{n}}{P_{50}^{\,n} + (pO_2)^{n}}$$
其中 $n$ 為 Hill 係數,$P_{50}$ 為達半飽和時的氧分壓。對血紅素 $n\approx 2.8$(介於 1 與 4 之間),反映正協同但非完全協同;若 $n=1$ 則退化為肌紅素那種雙曲線、無協同的單體結合。協同性的結構基礎是 T 態(緊張態,低親和力)與 R 態(鬆弛態,高親和力)間的構象切換:一個次單元結合氧後,次單元界面的鹽橋斷裂,整體向 R 態傾斜,提高其餘位點的親和力——MWC(Monod–Wyman–Changeux)齊變模型與 KNF(Koshland)序變模型即在描述此過程。
定量小範例:用 Hill 方程估計組織端的卸氧量
假設 $P_{50}=26\ \text{mmHg}$、$n=2.8$。
肺部($pO_2 = 100\ \text{mmHg}$):
$$Y_{\text{肺}} = \frac{100^{2.8}}{26^{2.8} + 100^{2.8}}$$
計算 $100^{2.8}=10^{2\times2.8}=10^{5.6}\approx 3.98\times10^{5}$;$26^{2.8}$:$\log_{10}26\approx1.415$,$2.8\times1.415=3.962$,故 $26^{2.8}\approx 9.16\times10^{3}$。
$$Y_{\text{肺}} \approx \frac{3.98\times10^{5}}{9.16\times10^{3}+3.98\times10^{5}} \approx 0.977$$
運動中的肌肉($pO_2 = 20\ \text{mmHg}$):$20^{2.8}$:$\log_{10}20\approx1.301$,$2.8\times1.301=3.643$,$20^{2.8}\approx 4.39\times10^{3}$。
$$Y_{\text{組織}} \approx \frac{4.39\times10^{3}}{9.16\times10^{3}+4.39\times10^{3}} \approx 0.324$$
卸氧分數 $=Y_{\text{肺}}-Y_{\text{組織}}\approx 0.977-0.324 = 0.653$,即約 65% 的氧在這段壓力差被釋放。值得注意:若血紅素是無協同的單體($n=1$),同樣兩點間僅釋放約 $0.79-0.43=0.36$,即 36%。協同性讓陡峭的 S 曲線把卸氧效率近乎翻倍——這正是四級結構的生理價值所在。
摺疊出錯的代價:聚集、伴護蛋白與蛋白質恆定
邊際穩定性也意味著摺疊容易失敗。未摺疊或錯摺疊的鏈會暴露疏水面,彼此黏聚形成不可逆的聚集體,乃至澱粉樣纖維(amyloid fibril)——其核心是富含 β 股的交叉-β 結構。阿茲海默症的 Aβ、帕金森症的 α-突觸核蛋白、第二型糖尿病的胰島澱粉樣多肽,皆屬此類「摺疊病」。
細胞為此演化出蛋白質恆定(proteostasis)網絡:Hsp70/Hsp40 結合暴露的疏水段防止過早聚集;GroEL/GroES(Hsp60)伴護蛋白桶提供一個被隔離的微環境讓鏈在其中重試摺疊;摺疊失敗者則經泛素—蛋白酶體系統或自噬清除。這些機制把熱力學上「可能但緩慢、易出錯」的摺疊,轉化為細胞內穩健、可控的過程。
深入探討(研究所視角)
當代結構生物學已從「單一靜態結構」邁向「構象系綜與動態」的描繪。冷凍電子顯微鏡(cryo-EM)在 2010 年代的「解析度革命」後,已能對大型、難結晶的寡聚複合體(如核糖體、剪接體、膜通道)解到近原子解析度,並透過 3D 分類捕捉同一樣本中共存的多種構象態,直接觀察別構切換的中間構象——這是傳統 X 光晶體學難以企及的。同時,氫—氘交換質譜(HDX-MS)以骨架醯胺氫的交換速率報告局部摺疊穩定性與溶劑暴露,提供殘基層級的動態「呼吸」資訊。
計算端的轉折同樣劇烈。AlphaFold2 與 RoseTTAFold 以演化耦合(共演化殘基對的接觸資訊,源自多序列比對)結合深度學習注意力機制,使序列到結構的預測精度在 CASP14(2020)達到可與實驗比擬的水準,AlphaFold3 進一步延伸到蛋白—配體、蛋白—核酸複合體。但須強調:這些工具預測的是最可能的天然態構象,而非摺疊「路徑」或自由能地景本身;理解摺疊動力學仍仰賴分子動力學(MD)模擬。Folding@home 等分散式運算與 Markov 狀態模型(MSM)的結合,能從大量短軌跡拼出毫秒級的摺疊動力學,量化中間態的居留時間與躍遷速率。
別構性的理論視角也在更新。除了經典 MWC/KNF 模型,現代觀點以「構象選擇」(conformational selection)補充「誘導契合」:未結合的蛋白本就在多個構象間動態取樣,配體只是選擇性地穩定其中之一,從而移動系綜的權重——這把別構性重新定義為對整個構象系綜的自由能再分配,甚至存在不涉及明顯構象變化、純由動態熵(側鏈運動幅度改變)驅動的別構途徑。
在系統與體學層次,蛋白質恆定不再被視為孤立的品管,而是一個耦合到細胞壓力反應的網絡:未摺疊蛋白反應(UPR)、熱休克反應(HSR)與整合壓力反應(ISR)共同調節伴護蛋白與降解機器的產能。單細胞蛋白體學(single-cell proteomics)與鄰近標記(如 BioID、APEX)正逐步揭示伴護蛋白—客戶蛋白互動網絡在不同細胞狀態下的異質性。值得注意的是,「天生無序蛋白」(intrinsically disordered proteins, IDPs)顛覆了「序列決定唯一結構」的傳統圖像——這類蛋白在生理條件下不摺疊成固定構象,卻能透過液—液相分離(LLPS)形成無膜胞器(如核仁、應激顆粒),以多價弱交互作用驅動凝聚。這提示摺疊的「終點」未必是單一致密球狀體,而可能是一個動態、可調控的凝聚相——當前生物物理與細胞生物學交會的最前沿之一。