
DNA 複製的分子機制與修復:保真度的工程學
從複製叉的分子機械到突變特徵——速度、能量與保真度如何在十億分之一的尺度上被同時優化
一個錯誤都不能多:複製的保真度賭注
每一次細胞分裂,人類細胞要把約 $6.4 \times 10^9$ 個鹼基對完整複製一份。如果用一支普通原子筆抄寫,抄到第十億個字才寫錯一個,已經堪稱神蹟;但細胞做得更極端——綜合錯誤率低到約 $10^{-9}$ 到 $10^{-10}$(每鹼基每次複製)。這個數字不是單一酵素的功勞,而是三道防線疊乘出來的結果:聚合酶本身的選擇性、$3' \to 5'$ 校讀外切活性,以及複製後的錯配修復(mismatch repair)。理解 DNA 複製,本質上是理解「速度、能量與保真度」如何在分子尺度上被同時優化。
DNA 複製是半保留(semiconservative)的——這是 Meselson–Stahl 用 $^{15}\text{N}$ 密度梯度離心在 1958 年釘死的結論。每股母鏈作為模板,合成出互補的新鏈。但「半保留」只是拓撲層面的描述,真正的工程難題藏在化學與幾何裡:兩股反向平行的鏈如何被一個只能單向($5' \to 3'$)合成的酵素同時複製?
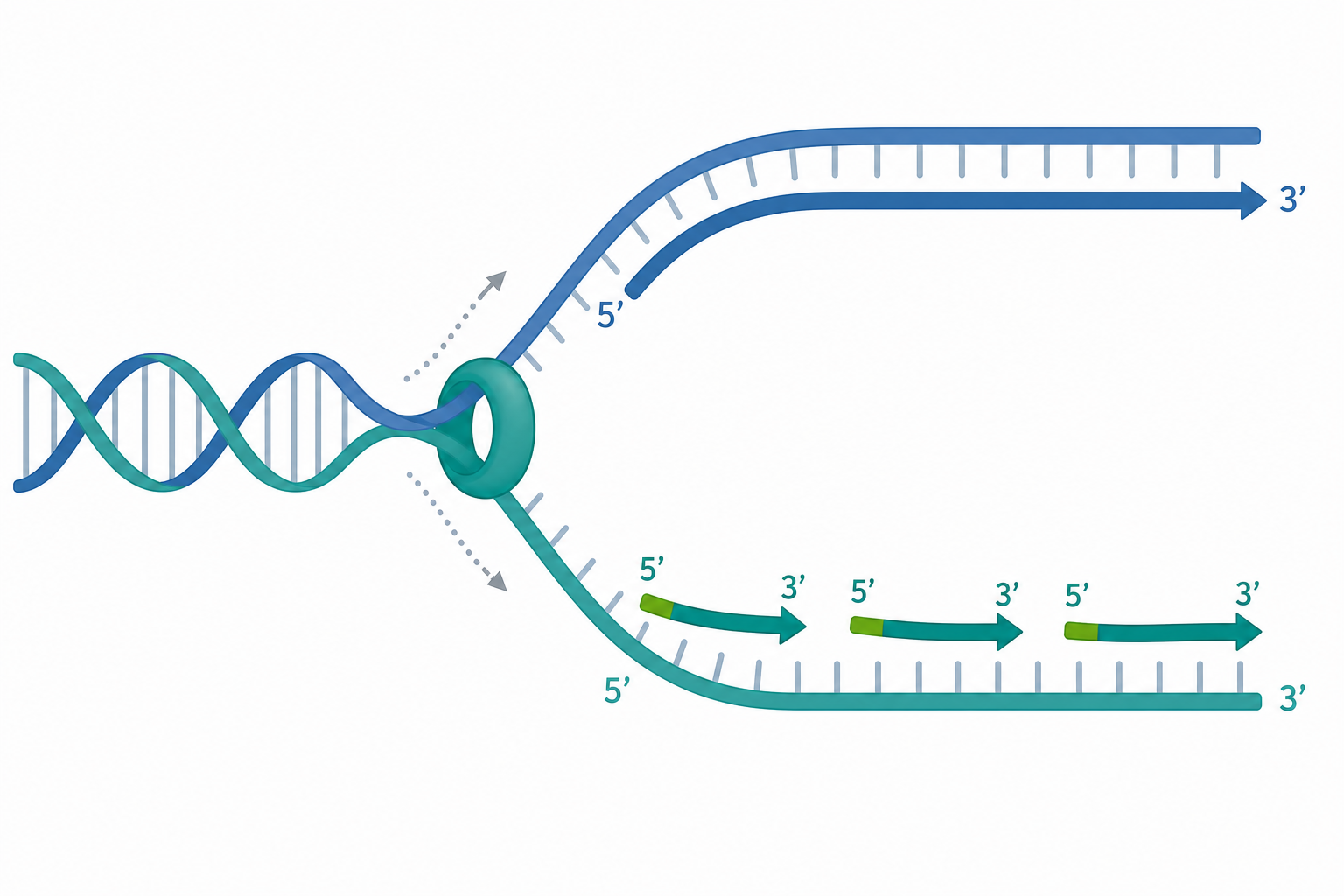
複製叉的分子機械:解旋、引子與不對稱合成
複製從特定的複製起點(origin)開始。在大腸桿菌中是單一的 oriC;真核生物則沿染色體散布數萬個起點,以確保 S 期能在數小時內完成。起點被識別後,解旋酶(helicase,原核為 DnaB、真核為 MCM2–7 六聚體環)利用 ATP 水解的自由能(每水解一個 ATP,$\Delta G \approx -30.5\ \text{kJ/mol}$,在細胞內實際更負)強行拆開雙股螺旋,產生兩條單鏈模板。單股 DNA 結合蛋白(SSB / RPA)立即包覆,防止其重新退火與二級結構形成。
聚合酶無法從零開始合成,必須有一段 RNA 引子(primer)。引子酶(primase,DnaG)合成約 10 個核苷酸的 RNA 短鏈提供游離的 $3'\text{-OH}$。隨後主力聚合酶(原核 Pol III、真核 Pol $\varepsilon$ 與 Pol $\delta$)以 dNTP 為基質延伸新鏈:
$$\text{DNA}_n + \text{dNTP} \longrightarrow \text{DNA}_{n+1} + \text{PP}_i$$
反應的熱力學驅動力很大一部分來自焦磷酸($\text{PP}_i$)被焦磷酸酶水解為兩個無機磷酸,把反應「拉」向合成方向,使縮合在生理條件下實質不可逆。
因為兩股反向平行,而聚合酶只能 $5' \to 3'$ 延伸,所以朝複製叉前進方向的「領先股(leading strand)」可連續合成;另一條「延遲股(lagging strand)」只能以反方向、片段化方式合成出岡崎片段(Okazaki fragments,原核約 1000–2000 nt、真核約 100–200 nt)。每個片段都需要新引子,事後由 RNase H 與 Pol I(原核)或 FEN1 + Pol $\delta$(真核)移除 RNA、補上 DNA,最後 DNA 連接酶(ligase)以 ATP 或 NAD$^+$ 為能源封合缺口。延遲股模板會被「迴圈」(trombone model)拉成環狀,讓同一個聚合酶全酶能與領先股協同前進。
保真度的數學:選擇性與校讀的疊乘
聚合酶選擇正確 dNTP 的本事,可用酵素動力學描述。摻入的「特異性常數」$k_{cat}/K_M$ 是判別正確與錯誤受質的核心量。保真度可近似為:
$$F = \frac{(k_{cat}/K_M)_{\text{correct}}}{(k_{cat}/K_M)_{\text{correct}} + (k_{cat}/K_M)_{\text{incorrect}}}$$
正確 Watson–Crick 配對與錯配在自由能上的差異看似只有幾 kJ/mol,但聚合酶活性中心透過幾何「誘導契合」放大了這個差異:只有正確配對能讓酶域閉合到催化所需的構型,使錯配的 $k_{cat}$ 驟降。單靠這層篩選,錯誤率約 $10^{-4}$ 到 $10^{-5}$。
第二道防線是 $3' \to 5'$ 外切校讀。當摻入錯誤鹼基,新生末端配對不良、延伸停滯,DNA 末端就有機會滑入外切活性中心被切除。校讀再壓低約 $10^{2}$ 倍錯誤率。第三道是複製後錯配修復,再壓 $10^{2}$ 到 $10^{3}$ 倍。三者相乘:$10^{-5} \times 10^{-2} \times 10^{-3} \approx 10^{-10}$,正好對上實測的生物保真度。
定量小範例。 假設某聚合酶對正確 dNTP 的 $k_{cat}/K_M = 1.0 \times 10^{7}\ \text{M}^{-1}\text{s}^{-1}$,對某錯配為 $1.0 \times 10^{3}\ \text{M}^{-1}\text{s}^{-1}$。則單純摻入階段的錯誤率為
$$P_{\text{err}} \approx \frac{1.0 \times 10^{3}}{1.0 \times 10^{7} + 1.0 \times 10^{3}} \approx 1.0 \times 10^{-4}.$$
若校讀再提供 $100$ 倍保護、錯配修復再提供 $1000$ 倍,總錯誤率約
$$1.0 \times 10^{-4} \times \tfrac{1}{100} \times \tfrac{1}{1000} = 1.0 \times 10^{-9}.$$
即每複製十億個鹼基才出現一個未修復的錯誤——這正是基因組穩定性的量化基礎。
修復網路:把損傷分門別類地拆解
複製只是基因組維護的一半,另一半是修復。DNA 每天承受水解(去嘌呤)、氧化、去胺、紫外線造成的嘧啶二聚體等成千上萬次損傷。細胞演化出多套互補的修復路徑:
- 錯配修復(MMR):辨識複製滑脫造成的鹼基錯配與小型插入/缺失環。關鍵在「如何分辨哪一條才是新鏈」——原核靠 Dam 甲基化的暫時半甲基化狀態(MutS–MutL–MutH 系統),真核則可能利用新鏈上未封合的缺口作為股別訊號。MMR 失能與 Lynch 症候群(遺傳性非息肉結直腸癌)直接相關。
- 鹼基切除修復(BER):由 DNA 醣苷酶移除受損的單一鹼基(如去胺後的尿嘧啶),形成無鹼基位點,再經 AP 內切酶、聚合酶補位與連接。
- 核苷酸切除修復(NER):移除造成螺旋扭曲的大型加合物與紫外線二聚體,以一段約 24–32 nt 的寡核苷酸整段切下。NER 缺陷導致著色性乾皮症(xeroderma pigmentosum),患者對日光極度敏感。
- 雙股斷裂修復:透過非同源末端接合(NHEJ)或較精確、需姊妹染色單體作模板的同源重組(HR)。BRCA1/2 屬於 HR 路徑,其失能是遺傳性乳癌與卵巢癌的核心機制,也是 PARP 抑制劑「合成致死」療法的標靶。
這些路徑並非各自為政,而是與複製叉緊密耦合:當複製叉撞上損傷或停滯,損傷耐受(DNA damage tolerance)與跨損傷合成(translesion synthesis)的低保真度聚合酶會接手,以「容許少量突變換取存活」的策略避免複製叉崩解。
深入探討(研究所視角)
主體把複製叉描述成有序的線性流程,但近十餘年的結構與單分子研究揭示了一幅遠更動態、隨機的圖像。
複製體的結構協同。 低溫電子顯微鏡(cryo-EM)已解析出真核 CMG 解旋酶(Cdc45–MCM–GINS)與 Pol $\varepsilon$ 的複合物結構,顯示解旋酶以 MCM 環包覆領先股模板、像分子馬達般沿單股移動。更出人意料的是,單分子螢光與磁鑷實驗顯示領先股與延遲股的合成並非嚴格同步:聚合酶會「停—走」(stop-and-go),岡崎片段起始與引子合成之間存在隨機等待,複製叉速度在毫秒尺度上劇烈波動。經典的「火車頭—煞車」協同被修正為更鬆散的隨機耦合,這對如何維持兩股拷貝數平衡提出了新的理論問題。
校讀的單分子動力學。 smFRET 已能直接觀測聚合酶活性位點與外切位點之間的 DNA 末端「轉移」(partitioning)。實驗顯示錯配並非總是立刻被切除,而是在兩個位點間動態分配,最終保真度是這個競爭過程的統計結果——把保真度從靜態的熱力學差異,提升為動力學分配問題。這也解釋了為何某些聚合酶突變能在不改變活性中心化學的前提下,僅透過改變構型轉換速率就大幅影響突變率。
體學與系統層面的突變足跡。 全基因組與單細胞定序催生了「突變特徵(mutational signatures)」分析:不同的內外在損傷與修復缺陷會在基因組留下可辨識的鹼基替換頻譜(如 NER 缺陷、MMR 缺陷、特定致癌物暴露各有指紋)。COSMIC 資料庫已歸納出數十種特徵,使我們能從一顆腫瘤的突變模式反推其曾經歷哪些修復路徑失效。單細胞層次更揭示:即使同一個體內,不同細胞累積的體細胞突變呈現顯著異質性,挑戰了「基因組在個體內近乎恆定」的舊假設。
複製壓力與系統生物學模型。 當 dNTP 庫不足、複製起點過度激發或遇到難複製區(脆弱位點),複製叉停滯形成「複製壓力(replication stress)」,是腫瘤發生的早期驅動力。ATR–CHK1 訊號網路扮演中央調控者,動態調節起點激發速率與叉的修復。研究者已用常微分方程與隨機模型描述起點激發的時空分布,量化「dormant origin」如何作為備援以救援停滯的叉。把分子事件升級成可預測的系統行為,正是當代複製研究與計算生物學交會的前沿——它讓「保真度」不再只是單酵素的化學參數,而是整個基因組在時空尺度上協調的湧現性質。