
細胞週期調控、檢查點與癌症:從分子開關到基因組失控
CDK–cyclin 動力學、三道檢查點與 p53 守護網路如何在腫瘤中失效
從「分子開關」看細胞為何分裂、何時停手
細胞分裂不是「時間到了就分」,而是一連串由蛋白質濃度與磷酸化狀態驅動的雙穩態切換(bistable switch)。直覺上,你可以把細胞週期想成一台只能單向前進、且在關鍵路口設有閘門的機器:每一道閘門都要確認「上一步真的做完了嗎?」才放行。當這些閘門的判讀邏輯被破壞,細胞就會在 DNA 損傷未修復、染色體未對齊的情況下強行前進——這正是癌症在分子層次的核心。本文聚焦調控網路的動力學、檢查點的訊號傳遞,以及它們如何在腫瘤中失效。
CDK–cyclin:以濃度與磷酸化驅動的相位引擎
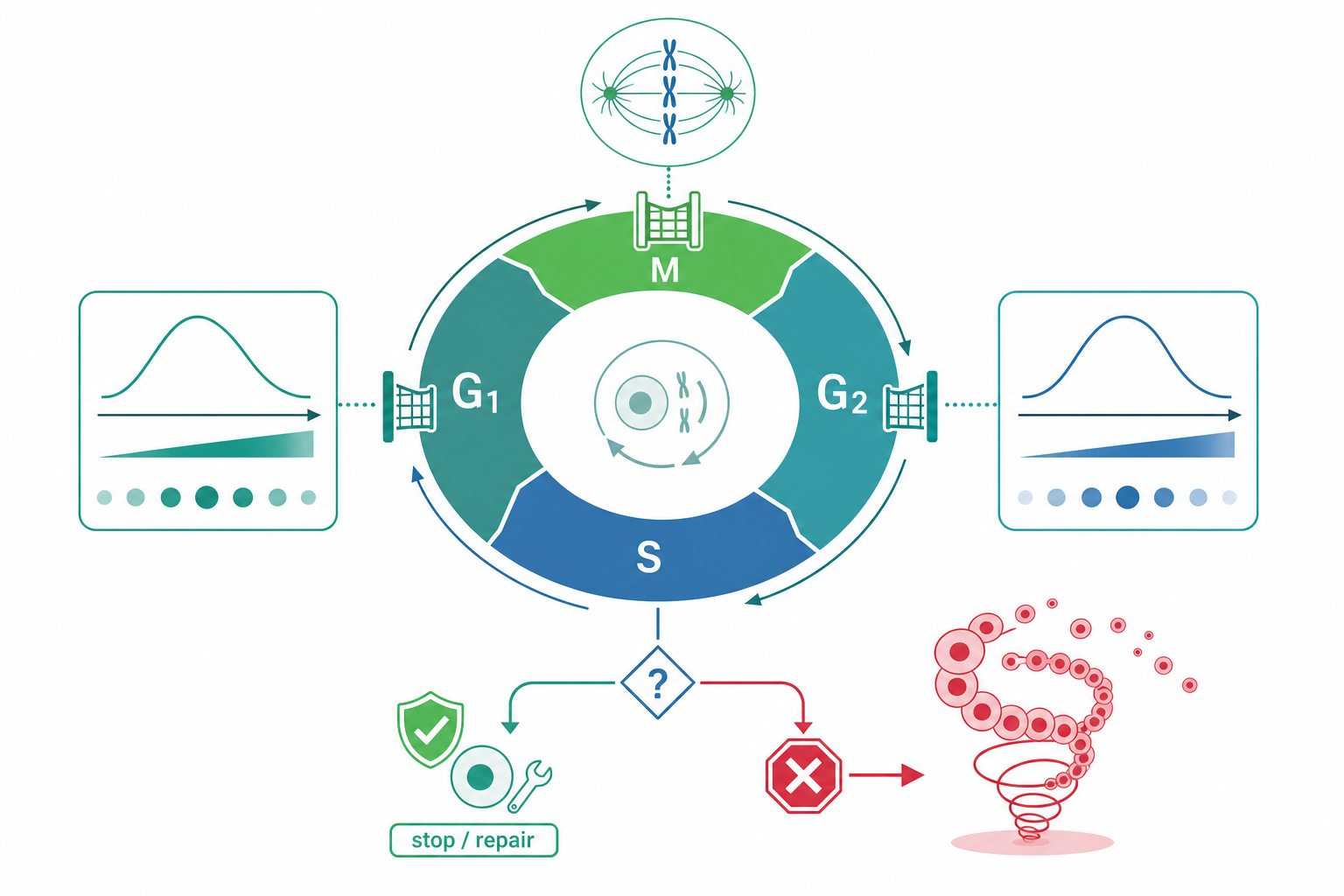
細胞週期的核心引擎是 cyclin-dependent kinase(CDK)與其調節次單元 cyclin。CDK 本身的激酶活性近乎為零,必須結合特定 cyclin 才被活化。不同相位由不同的 CDK–cyclin 複合體主導:G1 期的 CDK4/6–cyclin D 與 CDK2–cyclin E、S 期的 CDK2–cyclin A、M 期的 CDK1–cyclin B。
關鍵在於,cyclin 的濃度隨週期週期性合成與被泛素化降解(由 SCF 與 APC/C 兩大 E3 連接酶系統執行),而 CDK 的活性又被抑制性磷酸化(Wee1 在 Tyr15 加上磷酸基使其失活)與去磷酸化(Cdc25 磷酸酶移除該磷酸基使其活化)拉鋸。Wee1 與 Cdc25 之間存在雙重負回饋與正回饋,形成一個帶有遲滯(hysteresis)的超敏感開關——這解釋了為何 G2/M 轉換是「全有或全無」式的突發事件,而非緩慢漸進。
我們可用一個簡化的活化動力學描述去磷酸化反應速率。設活性 CDK1 對其受質的反應遵循 Michaelis–Menten 形式:
$$v = \frac{V_{\max}\,[S]}{K_m + [S]}$$
其中 $V_{\max}$ 正比於活性 CDK1 濃度。當 Cdc25(本身也是 CDK1 的受質)被活化後,會回頭活化更多 CDK1,使有效 $V_{\max}$ 隨自身產物上升——這個正回饋讓系統的劑量反應曲線從雙曲線變為陡峭的 S 形(Hill 係數 $n>1$):
$$\frac{v}{V_{\max}} = \frac{[S]^n}{K_{0.5}^{\,n} + [S]^n}$$
$n$ 越大,開關越「銳利」,這是檢查點能夠果斷決策、避免在臨界點來回猶豫的數學基礎。
三道檢查點:把「品質管制」寫進訊號網路
細胞週期有三個經典監測節點:
G1/S 檢查點(restriction point)。 這是細胞「決定是否進入分裂」的承諾點。Rb(retinoblastoma protein)在低磷酸化狀態下緊抓轉錄因子 E2F,抑制 S 期基因表現。當 CDK4/6–cyclin D 與 CDK2–cyclin E 逐步將 Rb 多位點磷酸化(hyperphosphorylation),E2F 被釋放、啟動 DNA 複製所需基因。p16^INK4a^ 抑制 CDK4/6,是此處的天然煞車。
G2/M 檢查點。 若 DNA 有損傷或複製未完成,ATM/ATR 激酶被招募至損傷位點,活化 Chk1/Chk2,後者磷酸化並抑制 Cdc25、同時穩定 Wee1,使 CDK1–cyclin B 無法被活化,細胞停在 G2 不進入有絲分裂。
紡錘體組裝檢查點(SAC)。 在中期,只要還有任一動粒(kinetochore)未被微管正確雙極附著,未附著的動粒就持續產生「等待訊號」——Mad2、BubR1 等組成 mitotic checkpoint complex(MCC),抑制 APC/C^Cdc20^,阻止 securin 與 cyclin B 被降解,因此姊妹染色分體無法分離。唯有全部動粒張力到位,訊號歸零,APC/C 才被解除抑制,觸發後期(anaphase)。
p53:基因組的守護者與決策樞紐
DNA 損傷訊號最終匯聚到轉錄因子 p53。平時 p53 被 MDM2 持續泛素化降解、濃度極低;損傷後 ATM/Chk2 磷酸化 p53 與 MDM2,切斷此降解,p53 濃度上升並啟動下游程式:誘導 p21(CDK 抑制因子,執行週期停滯)、修復基因,若損傷過重則啟動凋亡(PUMA、BAX)。p53 並非簡單開關,而是以脈衝式(pulsatile)動態編碼損傷嚴重度——脈衝次數與幅度決定細胞「修復後續活」或「走向凋亡」。
定量小範例:抑制劑如何改變半飽和點。 假設某 CDK 抑制劑為競爭性抑制(與受質競爭活性位),其表觀 $K_m$ 變為 $K_m^{app}=K_m(1+[I]/K_i)$。設 $K_m=2\ \mu M$、$K_i=0.5\ \mu M$、抑制劑濃度 $[I]=1.5\ \mu M$:
$$K_m^{app}=2\times\left(1+\frac{1.5}{0.5}\right)=2\times4=8\ \mu M$$
若受質濃度固定在 $[S]=2\ \mu M$,加抑制劑前反應速率 $v/V_{\max}=2/(2+2)=0.50$;加抑制劑後 $v/V_{\max}=2/(8+2)=0.20$。也就是說在不改變 $V_{\max}$ 的前提下,活性下降至原本的 40%。這說明 CDK4/6 抑制劑(如臨床上的 palbociclib 類藥物)為何能在不立即殺死細胞的情況下,把 G1/S 通量壓到使腫瘤停滯的水準。
癌症:檢查點失效的累積結果
癌症本質上是這套調控網路被多重突變「鬆綁」。Hanahan 與 Weinberg 提出的腫瘤特徵中,至少有兩項直接對應本文機制:持續的增殖訊號(如 cyclin D 過量表現、Rb 失活、p16 缺失使 G1/S 閘門失靈)與規避生長抑制(p53 突變使損傷不再觸發停滯或凋亡——p53 是人類腫瘤中最常突變的基因)。
更深一層,檢查點失效會導致基因組不穩定性:SAC 鬆弛造成染色體分離錯誤,產生非整倍體(aneuploidy);G2/M 把關失靈讓帶斷裂的染色體進入分裂,引發染色體碎裂(chromothripsis)。這種不穩定本身又加速更多致癌突變累積,形成惡性循環。正因如此,許多化療藥物(紡錘體毒素、DNA 損傷劑)的策略,是刻意製造 SAC 或 DNA 損傷檢查點難以承受的壓力,迫使本已脆弱的腫瘤細胞走向有絲分裂災難(mitotic catastrophe)。
深入探討(研究所視角)
結構生物學:CDK 活化的構象幾何。 CDK 的活化不只是「結合 cyclin」這麼簡單。冷凍電鏡與晶體結構顯示,cyclin 結合會重新排列 CDK 的 PSTAIRE α 螺旋,使催化關鍵的麩胺酸正確面向 ATP 的 γ-磷酸;接著 CDK-activating kinase(CAK,即 CDK7–cyclin H–MAT1)磷酸化活化環(T-loop)上的保守蘇胺酸(CDK1 為 Thr161、CDK2 為 Thr160),使 T-loop 翻出活性位、形成完整受質結合面。這種「兩步活化」(結合誘導 + 磷酸化鎖定)是動力學遲滯的結構根源。p27^Kip1^ 等 CIP/KIP 抑制因子則以延伸鏈插入 ATP 結合裂縫,直接阻斷催化——這也是為何同一個 p27 在不同磷酸化狀態下可從抑制劑轉為組裝因子,呈現「劑量與修飾依賴」的雙面性。
單細胞與體學:揭示群體平均掩蓋的決策異質性。 傳統同步化細胞群測得的是平均行為,但單細胞活細胞影像(如 CDK2 活性的 FRET/螢光報告子、APC/C 受質報告子)揭示了一個關鍵發現:細胞在有絲分裂結束時並非一致進入下一個 G1,而是依母細胞累積的損傷與 p21 濃度,分岔為「CDK2-increasing(立即承諾再分裂)」與「CDK2-low(進入暫時靜止)」兩群。這種出生時就決定的命運分流,無法從群體平均推得,凸顯了單細胞解析度對理解 restriction point 的不可取代性。結合單細胞 RNA-seq 與 ATAC-seq,研究者更能把週期相位、轉錄狀態與染色質可及性對齊到擬時序(pseudotime)軌跡上。
系統生物學:把檢查點寫成可預測的動力學模型。 Novak 與 Tyson 等人以常微分方程組(ODE)建構 CDK–cyclin–Cdc25–Wee1 網路,成功預測 G2/M 轉換的雙穩態與遲滯區間,並以實驗(Xenopus 卵萃取液)驗證了「活化閾值高於去活化閾值」的記憶效應。這類模型把分子交互作用形式化為相位平面上的鞍結點分岔(saddle-node bifurcation),使「為何系統一旦過閾就無法回頭」成為可計算的命題。近年更將 p53 脈衝、ATR–Chk1 回饋納入混合(hybrid)隨機模型,解釋為何相同劑量輻射下個別細胞命運仍呈機率性分布——把「噪音」從干擾項提升為決策的功能性要素。
前沿治療機制。 合成致死(synthetic lethality)概念在此格外深刻:BRCA 突變細胞已喪失同源重組修復,再以 PARP 抑制劑阻斷單股斷裂修復,便迫使其在複製時累積無法修復的雙股斷裂而死亡——這是「利用一道檢查點/修復路徑已失效,去攻擊其備援路徑」的精準策略。WEE1 抑制劑、ATR 抑制劑亦循同理:對已失去 p53(G1 把關)的腫瘤,撤掉其僅存的 G2/M 後備閘門,使其在複製壓力下崩潰。這些都建立在本文所述檢查點網路的精確機制圖譜之上。