
為什麼放大一萬倍就失敗?化學工程的「尺度」之戰
以無因次數、停留時間分布與相似性放大法則,解釋為何同一個化學反應在燒杯成功、在工業槽失敗,並連結程序最佳化。
為什麼放大一萬倍就失敗?化學工程的「尺度」之戰
入門篇留下了一句沒交代清楚的話:很多在燒杯裡漂亮的反應,放大一萬倍後就因為散熱不及、混合不均或分離成本太高而徹底失敗。這句話聽起來像經驗談,但它背後藏著化學工程最深刻、也最反直覺的一個事實——當你把一個系統等比例放大時,不同的物理過程並不會以相同的比例跟著放大。
考慮一個簡單的問題。假設你把實驗室的球形反應槽直徑放大十倍,體積(也就是反應物的量、放熱的量)會增加 $10^3 = 1000$ 倍,但散熱要靠的表面積只增加 $10^2 = 100$ 倍。換句話說,產熱以體積($L^3$)的速率成長,散熱卻只以表面積($L^2$)的速率成長。放大十倍後,每單位散熱面積要承擔的熱量變成了十倍。一個在燒杯裡溫溫和和的放熱反應,在工業槽裡可能因為來不及散熱而急速升溫、反應加速、放出更多熱——這個正回饋一旦失控,就是化工廠最致命的熱失控(thermal runaway)。
這就是本文要談的核心:化學工程不是「把化學變大」,而是「在不同尺度上,重新平衡彼此競爭的物理過程速率」。理解這件事,你才真正理解了化工為何是一門獨立的工程學科,而不是「大號的化學」。
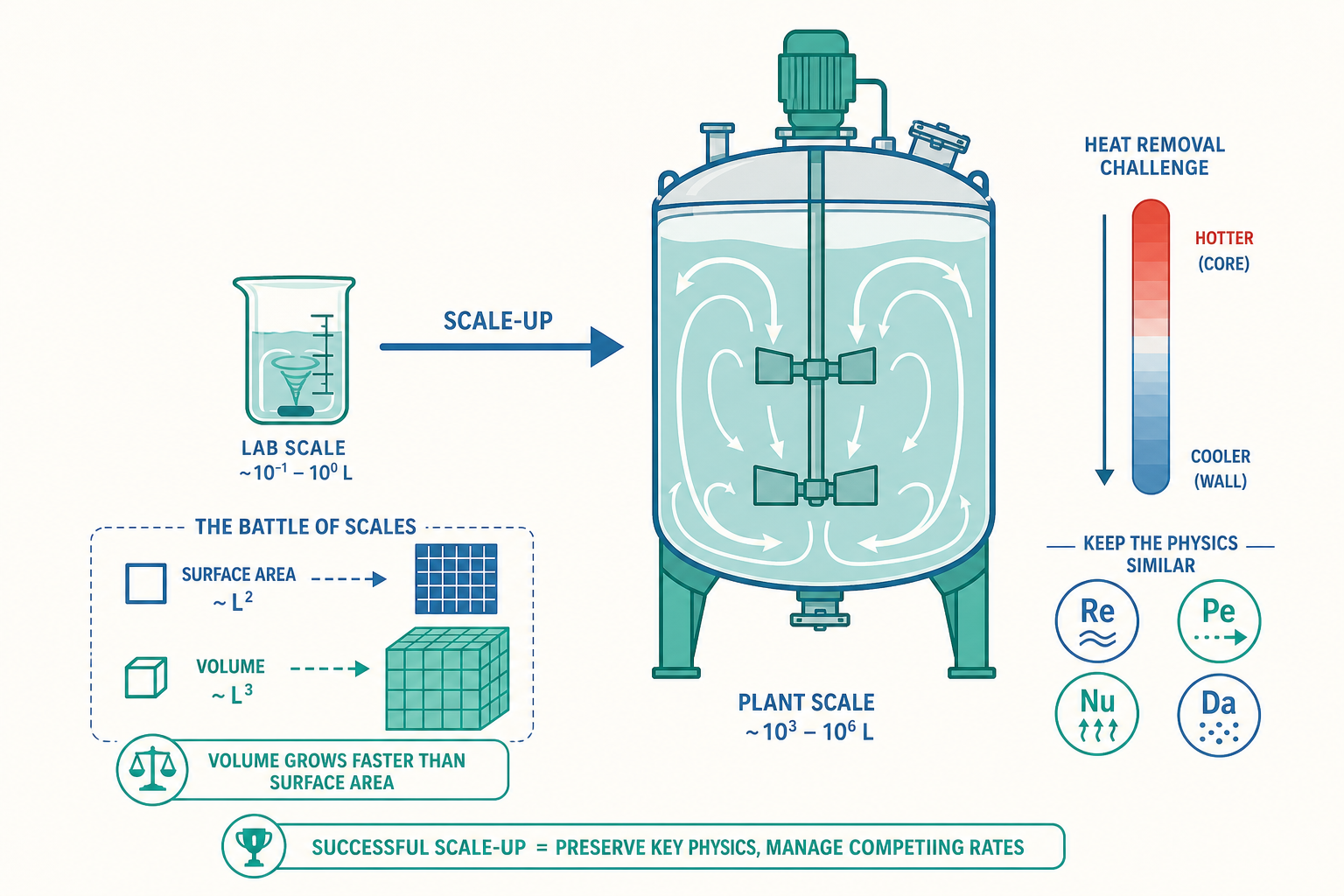
尺度的語言:無因次數(dimensionless numbers)
入門篇用質能平衡算「總量」、用傅立葉與菲克定律算「速率」。但當我們要比較兩個速率誰快誰慢——例如「反應快還是擴散快」、「對流快還是傳導快」——最有力的工具是無因次數(dimensionless number)。它的本質是把兩個競爭過程的特徵速率(或特徵時間)相除,得到一個沒有單位的純數。這個數的大小,就決定了系統由誰主導。
最著名的是雷諾數(Reynolds number, $Re$),它衡量慣性力與黏滯力的比值:
$$Re = \frac{\rho u L}{\mu}$$
其中 $\rho$ 是密度、$u$ 是特徵流速、$L$ 是特徵長度、$\mu$ 是黏度。$Re$ 小時黏滯力主導,流動平順(層流);$Re$ 大時慣性主導,流動翻騰混亂(紊流)。圓管內大約 $Re > 4000$ 就進入紊流。這個數為什麼重要?因為紊流的混合與傳熱效率遠高於層流——所以同一支泵、同一根管,光是改變流速跨過臨界 $Re$,傳熱係數可能跳升數倍。
與「反應 vs. 傳輸」直接相關的是丹克勒數(Damköhler number, $Da$),它是反應速率與輸送速率的比值。以一級反應、對流輸送為例:
$$Da = \frac{\text{反應速率}}{\text{對流速率}} = \frac{k \, \tau}{1} = k\,\tau$$
其中 $k$ 是反應速率常數、$\tau$ 是物料在系統內的停留時間(residence time)。$Da \ll 1$ 表示反應太慢、物料還沒反應就被沖走,轉化率低;$Da \gg 1$ 表示反應極快、瞬間就反應完,此時系統的瓶頸反而是「能不能把反應物送到夠快」——也就是輸送限制(transport-limited)而非反應限制。
這個觀念解釋了入門篇那句「混合不均」的失敗。當 $Da$ 很大,反應在反應物剛接觸的瞬間就完成,於是整個程序的快慢不再由化學決定,而由「混合把反應物送到一起的速度」決定。燒杯靠磁攪拌子就能瞬間混勻,工業槽裡上萬公升的液體卻可能有死角、有混合死區——同一個化學,在兩個尺度上落在 $Da$ 數軸的不同主導區,行為完全不同。
停留時間分布:理想與真實的鴻溝
入門篇的物料平衡假設每個單元都是「黑盒子」,流進等於流出。但真實反應器內部,不是每一個分子都待一樣久。有的分子走捷徑很快溜出去(短路,short-circuiting),有的卡在死角繞很久(死區,dead zone)。描述這件事的工具,是停留時間分布(Residence Time Distribution, RTD)。
化工教科書裡有兩個極端的理想模型。一個是連續攪拌槽反應器(CSTR),假設槽內瞬間完全混合,所以出口濃度等於槽內濃度,任一分子的停留時間服從指數分布。另一個是塞流反應器(Plug Flow Reactor, PFR),假設流體像一根活塞往前推,每個分子停留時間完全相同。對同樣的反應與相同的平均停留時間 $\tau = V / \dot{v}$($V$ 是反應器體積、$\dot{v}$ 是體積流率),這兩種理想反應器給出的轉化率並不相同——對正級數反應,PFR 通常比 CSTR 達到更高的轉化率。
真實反應器落在這兩個理想之間。工程師用一個示蹤劑脈衝(tracer pulse)——在入口瞬間注入一小撮可偵測的物質,再在出口量測它隨時間流出的濃度曲線 $C(t)$——就能反推出 RTD 函數:
$$E(t) = \frac{C(t)}{\displaystyle\int_0^\infty C(t)\,dt}$$
$E(t)\,dt$ 代表停留時間落在 $t$ 到 $t+dt$ 之間的物料分率,且 $\int_0^\infty E(t)\,dt = 1$。平均停留時間則是它的一階矩 $\bar{t} = \int_0^\infty t\,E(t)\,dt$。如果量到的 $\bar{t}$ 明顯小於理論的 $\tau = V/\dot{v}$,就代表有死區把有效體積吃掉了。RTD 是把入門篇那個理想「黑盒子」打開、診斷它到底偏離理想多遠的聽診器。
看一個例子
讓我們把丹克勒數與停留時間串起來,量化「為什麼放大會掉轉化率」。
問題:某一級液相反應 $A \to B$,反應速率常數 $k = 0.5 \text{ min}^{-1}$。我們用一個塞流反應器(PFR)操作。實驗室小試的停留時間 $\tau_1 = 6$ 分鐘。後來放大生產時,因為產量需求,體積流率提高,使得停留時間縮短為 $\tau_2 = 2$ 分鐘(反應器體積來不及等比例加大)。問轉化率各是多少?
PFR 一級反應的轉化率公式(由 PFR 設計方程 $\tau = \int_0^{X} \frac{dX}{k(1-X)}$ 積分而得):
$$X = 1 - e^{-k\tau} = 1 - e^{-Da}$$
這裡 $Da = k\tau$ 正是丹克勒數。
小試:$Da_1 = 0.5 \times 6 = 3.0$
$$X_1 = 1 - e^{-3.0} = 1 - 0.0498 \approx 0.950$$
轉化率約 95%,漂亮。
放大後:$Da_2 = 0.5 \times 2 = 1.0$
$$X_2 = 1 - e^{-1.0} = 1 - 0.368 \approx 0.632$$
轉化率掉到 63%。同樣的化學、同樣的溫度、同樣的反應速率常數,只因為停留時間縮短了三分之二,轉化率就從 95% 崩到 63%。剩下的 37% 未反應原料要嘛浪費、要嘛得增設回收單元——這就是「化學沒變、工程卻失敗」的精準量化。要救回轉化率,工程師必須回頭把反應器體積加大,讓 $\tau$ 回到夠大、$Da$ 回到夠高的區間。這正是放大設計(scale-up)的核心攻防。
放大的法則:保持什麼不變?
既然等比例放大會破壞各過程速率的平衡,那放大設計到底在做什麼?答案是:選定一個最關鍵的無因次數或特徵量,在放大時想辦法維持它不變,而接受其他次要量隨之改變。這稱為相似性放大(similarity scale-up)。
問題在於,你往往無法同時維持所有量不變。以攪拌槽放大為例,常見的兩種策略:
- 維持單位體積功率(power per unit volume, $P/V$)不變——適合需要良好分散、混合的程序。
- 維持葉輪尖端速度(tip speed, $\pi N D$)不變——適合對剪切力敏感(如細胞培養、易碎晶體)的程序。
數學上可以證明,這兩個目標在放大時是互相衝突的:固定 $P/V$ 會讓尖端速度上升,固定尖端速度又會讓 $P/V$ 下降。換句話說,沒有一種放大能讓所有現象同時保持相似,工程師必須判斷哪一個現象是這個程序的速率瓶頸,犧牲其他來保住它。這種「不可能全贏、只能抓主要矛盾」的取捨,正是工程判斷(engineering judgment)的精髓,也是它與「按公式算」的最大差別。
這也回應了一個常見迷思:放大不是「把圖紙乘以一個係數」。一座千噸級反應器,往往要先在實驗室(毫升級)建立反應動力學,再到中試廠(pilot plant,公斤到噸級)驗證輸送與混合行為,最後才敢蓋商業廠。每跨一個尺度,主導的物理可能就換人——這就是為什麼化工被稱為一門多尺度(multiscale)的學科。
把尺度寫進方程:無因次化
無因次數不只是經驗比值,它會自然地從控制方程中「掉出來」。我們把入門篇那條對流–擴散–反應方程拿來無因次化,就能親眼看到 $Da$ 與另一個無因次數(Péclet 數)如何主宰系統。
考慮穩態一維對流–擴散–反應:
$$u\frac{dC}{dx} = D\frac{d^2 C}{dx^2} - kC$$
引入無因次變數:$C^* = C/C_0$(以入口濃度 $C_0$ 正規化)、$x^* = x/L$(以系統長度 $L$ 正規化)。代入並整理,方程變成:
$$\frac{dC^*}{dx^*} = \frac{1}{Pe}\frac{d^2 C^*}{dx^{*2}} - Da\,C^*$$
其中冒出來兩個純數:
$$Pe = \frac{uL}{D} \quad(\text{Péclet 數,對流 vs. 擴散}), \qquad Da = \frac{kL}{u} \quad(\text{丹克勒數,反應 vs. 對流})$$
這個式子的威力在於:整個系統的行為,不再取決於 $u$、$L$、$D$、$k$ 這四個有單位的參數各自的大小,而只取決於 $Pe$ 與 $Da$ 這兩個無因次組合。兩座尺寸天差地遠的反應器,只要 $Pe$ 與 $Da$ 相同,它們的無因次濃度分布 $C^*(x^*)$ 就完全一樣——這正是相似性放大的理論基礎。$Pe \to \infty$(擴散可忽略)時方程退化成塞流模型,$Pe \to 0$(擴散主導、瞬間混勻)時退化成完全混合模型。一條方程,靠兩個無因次數,就把所有尺度的反應器連成了一張連續的地圖。
重點回顧
- 體積與表面積的尺度律不同($L^3$ vs. $L^2$)是放大失敗的根源:產熱、產物量隨體積成長,但散熱、傳質的面積只隨平方成長,放大後散熱與混合相對「跟不上」。
- 無因次數是比較競爭過程速率的語言:雷諾數($Re$)分層流/紊流、丹克勒數($Da=k\tau$)分反應限制/輸送限制、Péclet 數($Pe$)分對流/擴散主導。
- 停留時間分布(RTD)揭露真實反應器偏離理想(CSTR/PFR)多遠,用示蹤劑脈衝量測 $E(t)$ 即可診斷死區與短路。
- 放大不能維持所有相似性:固定 $P/V$ 與固定尖端速度互相衝突,工程師必須辨識速率瓶頸、抓主要矛盾,這是工程判斷的核心。
- 無因次化讓控制方程「吐出」主導的無因次數,證明只要 $Pe$、$Da$ 相同,不同尺度的系統行為一致——這就是相似性放大的數學地基。
深入探討(研究所視角)
本文用單一無因次數描述「主導現象」,是一種一維、平均化的視角。真實的放大問題往往是多個無因次數同時起作用、且彼此耦合的高維問題。研究所層級的反應工程,會把對流–擴散–反應方程與動量方程(Navier–Stokes)、能量方程完全耦合,用計算流體力學(CFD)在三維幾何上求解,直接「算出」攪拌槽裡的混合死區、溫度熱點與局部 $Da$ 分布,而非依賴經驗放大係數。
更前沿的方向是把放大從「靠經驗逐級驗證」轉為「靠模型直接最佳化」。一個現代化工設計問題會被寫成一個受約束最佳化:
$$\min_{\mathbf{d}} \; C_{\text{total}}(\mathbf{d}) \quad \text{s.t.} \quad \mathbf{g}(\mathbf{d}) = 0,\; \mathbf{h}(\mathbf{d}) \le 0$$
其中決策變數 $\mathbf{d}$ 是反應器尺寸、操作溫度、流率等;等式約束 $\mathbf{g}$ 是質能平衡與輸送方程(可能是一整組偏微分方程離散後的代數式);不等式約束 $\mathbf{h}$ 涵蓋安全溫度上限、轉化率下限、環保排放法規。這正好接回 Uedu 的優化學主題——程序放大本質上是在「物理定律不可違反」的可行域裡,尋找成本最低的那一點。
當代的難點在於,高保真的 CFD 或動力學模型計算太貴,無法直接塞進最佳化迴圈反覆求解。於是研究者用機器學習訓練代理模型(surrogate model):以少量昂貴的高保真模擬資料,學一個快速可微的近似映射,再用它在最佳化器裡高速搜尋;並結合不確定性量化(uncertainty quantification),在動力學參數本身有誤差時,仍能設計出在整個參數分布上都安全可靠的程序(robust design)。值得強調的是,這些花俏的資料驅動方法,全都站在本文這套無因次分析與輸送平衡的基本功之上——機器學習可以幫你內插,卻無法替你判斷「哪個無因次數才是這個程序的主要矛盾」。那一步的物理洞察,至今仍是化學工程師不可被取代的價值。